本文作者:孙苏赟
在之前,我们了解了醇的一系列转化,例如将醇的羟基转化成卤原子和磺酸酯。这些物种和羟基比起来都是不错的离去基团,因此可以用于发生取代反应。
- 烷基卤代烃和磺酸酯的亲核取代反应 (SN2)
在饱和碳原子上,亲核取代反应可以相对容易地发生,以此可以作为生成杂原子官能团的方法,利用这个原理甚至可以实现新得碳-碳键的形成。亲核取代反应可以被很多因素影响,例如例如基团,亲核试剂,反应的条件(例如溶剂和温度)和底物自身的性质。
- 溶剂:极性溶剂对反应有利,例如DMSO,DMF等。它们可以提高反应速率,并且可以高效的溶剂离子化合物。此外,相转移催化剂可以用于解决溶解性的问题。
- 离去基团的离去性质:ROTf >> ROMs ~ ROTs ~ RI > RBr > RCl
- 底物:苄基,烯丙基 > Me- > 1o烷基 > 2o烷基;由于位阻的原因,3o烷基的卤原子和磺酸酯一般不可发生亲核取代反应,通常发生的是消除反应。
(1) 氰基

这是氰基取代的非常有用的一种方法。生成得到的氰基又可以进一步水解得到羧基,而羧酸又可以不同程度的被还原得到醛和醇。反应的条件一般是醇和水作为混合溶剂,同时反应需要高温,也可以使用非质子型极性溶剂,例如DMSO,DMF。
(2) 叠氮化合物

利用卤原子和磺酸酯转化成叠氮基团是将醇转化成胺的一种非常有效而间接的方法,之后利用烷基膦进行Staudinger反应即可转化成氨基,最常用的是PPh3:
它的反应机理是:
之前也提到过,叠氮化合物也可以由醇通过Mitsunobu反应得到:

(3) 氧亲核试剂

甲基和苄基底物是最适合利用此方法合成醚类化合物,其他基团可能会导致存在副反应,例如底物的消除,等。
(4) 氮亲核试剂(Gabriel合成法)

Gabriel合成法的发明是为了克服胺合成中难以避免的过度烷基化的问题,氮不足的是反应需要分两步进行,其中一步为联氨作用下的碎裂反应。此外Mitsunobu反应和还原胺化反应也可以达到不错的效果。其中,氮亲核试剂还有其他的替代:
- 芳香卤化物和磺酸酯
(1) 亲核芳香取代反应 (SNAr)
这是一类非常有效的方法,氮反应条件常常比较苛刻,只有特定的芳环才可以发生此类反应,例如芳环上邻位和/或对位有强吸电基团。
(2) Ullmann偶联
Ullmann反应的经典应用是用于芳香醚的合成,反应在高温下使用Cu(I)试剂作为催化试剂,其中ArO-Cu(I)可能是反应中经历的中间体。此外胺也可作为反应的组分,然而这个转化现在常常由Buchward-Hartwig反应实现。例如[1]:
(2) Buchward-Hartwig偶联 [2-3]
在Buchward-Hartwig反应中,醇,酚和其他几种氮亲核试剂可以作为反应的一个组分,另一个组分则是芳基卤代物或芳基磺酸酯。反应条件温和高效,需要Pd或Ni作为催化剂参与其中。
反应大致过程如下:
几个例子:
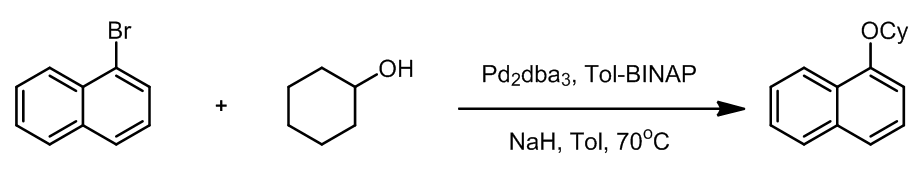
- 芳基-2o烷基偶联 [4]
- 杂环芳烃的偶联 [5]
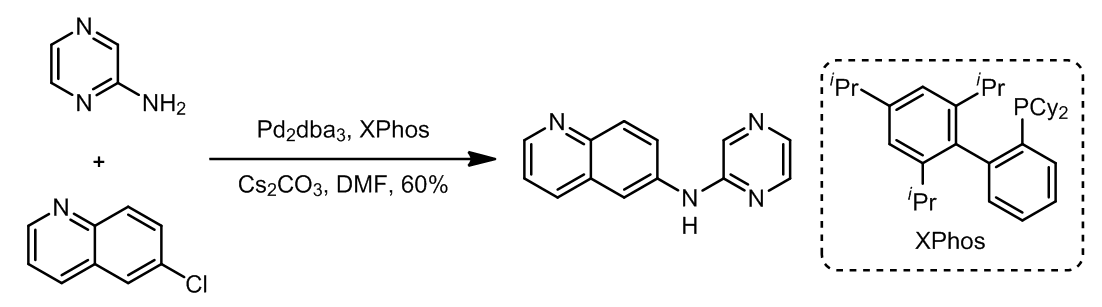
- 杂环芳烃的偶联[5]
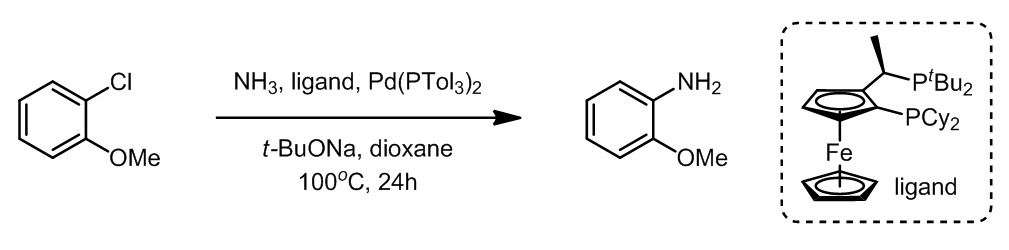 由于氨和金属Pd的强烈的相互作用导致氨直接参与偶联反应是非常困难的,因此反应中需要使用过量的氨。
由于氨和金属Pd的强烈的相互作用导致氨直接参与偶联反应是非常困难的,因此反应中需要使用过量的氨。



(3) Chan-Lam偶联

Chan和Evans课题组在1998年分别报道了醋酸酮催化的芳基和杂环芳香硼酸和芳基的偶联反应,反应底物包括酚,硫酚,芳胺,等。一个例子[6]: REFERENCES
REFERENCES
- Am. Chem. Soc.1997, 119, 43, 10539-10540, doi: 10.1021/ja971901j
- Chem. Int. Ed., 1998, 37, 2046, doi:10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L
- Chem. Int. Ed., 2008, 47, 6338, doi: 10.1002/anie.200800497
- Am. Chem. Soc.1997, 119, 143395-3396, doi: 10.1021/ja9640152
- Chem. Int. Ed., 2006, 45, 6523, doi: 10.1002/anie.200601612
- Tetrahedron , 1998, 39, 2937, doi: 10.1016/S0040-4039(98)00502-4
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者孙苏赟
一、碘代反应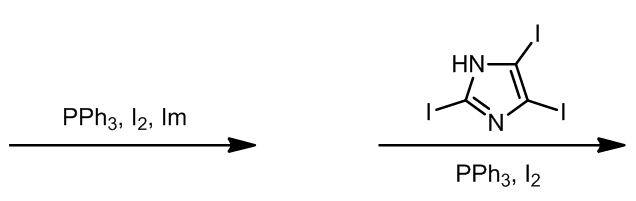

这种组合是最常用的碘代的方法,例如[1]:

此方法也常用于糖化学中:

此外还有一些不太常用的方法:

二、磺化反应
在有机合成中,磺酸酯作为效果还不错的离去基团,在有机合成中常作为卤素原子的替代,实现一系列的转化,较常用的磺酸酯的有甲磺酸酯 (Ms),三氟甲磺酸酯 (Tf)和对苯甲磺酸酯 (Ts):

离去基团的离去能力:ROTf >> ROMs ~ ROTs ~ RI > RBr > RCl

其中,(a) 是最常用的Ts磺化的方法,但是烯丙基和三级烷基炔非常不稳定,并且活性非常高,因此比较难制备提纯得到纯净物;(b) 是Ms磺化最常用的方法;(c) 用于大位阻的醇化合物;(d) 途径中使用三氟甲磺酸酐来制备Tf磺酸酯,二级醇的反应性更好,又因为可以发生SN2,因此在糖化学中非常常用。
这里是两个例子:
(1) 邻二醇的磺化-取代反应[2]:

(2) 二级醇的反应[3]:

三、磷酸酯化反应
磷酸酯在有机合成中不是非常常用,但是有时却是非常有效的反应试剂,例如Newhouse课题组开发的环酮的脱氢反应[4]:

但是磷酸酯的合成是很简单的:

反应过程中磷的手性会发生翻转。
四、低聚核苷酸的合成(oligonucleotide synthesis) [5, 6]
在低聚核苷酸的合成研究初期,磷化合物是其研究的核心。其中一个非常主要的问题就是分子中P-O–的结构的导致反应的选择性问题。这个问题也导致了后来的磷酸基团选择性活化的问题。
然而,在此之后Marrin Carruthers开发了一种亚磷酰胺的方法可以解决以上的难题。亚磷酰胺是一种很稳定的磷酸化试剂。而到了今天,这类反应常常以固相反应的形式在硅胶上进行。其中,腺嘌呤核苷和胞嘧啶上的氨基使用苯基作为保护基,鸟嘌呤核苷上的氨基可使用异丙基保护,而胸腺嘧啶的氨基则不需要保护基。
Carruthers开发的方法基本如下:


REFERENCES
- J. Am. Chem. Soc.1999, 7423-7424, DOI: 10.1021/ja991538b
- Org. Lett.1999, 11, 71-74,DOI: 10.1021/ol9905506
- Org. Lett.1999, 13, 499-502, DOI: 10.1021/ol990102y
- Org. Lett. 2018, 20, 684–687, DOI: acs.orglett.7b03818
- Science 1985 230 281, DOI: 1126/science.3863253
- Tetrahedron 1992, 48, 2223, DOI: 1016/S0040-4020(01)88752-4
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者孙苏赟
一、氯代反应
(1). SOCl2或PCl3/PCl5
这是最常见的醇的氯化的方法。
反应机理:

使用氯化磷作为氯源的反应机理是与此类似的。
(2) Ph3P/NCS (or (Cl3C)2CO, or CCl4)
这个方法中生成的膦氧化物中间体是一个很好的离去基团,对于底物具有很广泛的适应性,并且烯丙基醇的反应不会发生重排过程。
反应机理:

上边的是六氯丙酮的反应机理,NCS和CCl4的机理也是类似的:

(3) MeLi then TsCl/LiCl:这个方法中使用烷基锂试剂作为碱,会形成氧负离子,适用于大位阻的醇化合物。
(4) TsCl/NaCl

这个方法对于烯丙基醇也具有优势,具有较好的区域选择性:

在这个例子中,避免了烯丙基的重排,可以使烯丙基醇单一的转化成烯丙基氯化合物:

(4) 此外还有几种相对不是很常用的方法:

使用DMS和DCS搭配,可以对烯丙醇和苄醇有很好的选择性,并且其他饱和醇类在此条件先呈现惰性。
第二个方法其实是和Mitsuobu反应类似的,相同地,反应中会导致手性中心的反转。这个方法对于烯丙基醇和饱和醇都是适用的。
两个例子:
- 这是Meyer课题组开发的一种方法,这种方法对于四取代的烯丙醇比较有效[1]。

2. 磷试剂根据实际情况可能需要进行筛选[2]:

二、溴代反应

以上的方法在一级醇上会比二级醇上的效果好一些。反应过程是通过形成溴-膦正离子物种中间体进行的:

例如:
(1) 溴代过程中实现构型翻转[3]:

(2) 在Li+的协助下,溴负离子亲核进攻完成溴代反应[4]:

REFERENCES
- J. Org. Chem.199560247796-7814,DOI: 10.1021/jo00129a021
- J. Am. Chem. Soc.2002, 124, 15196-15197, DOI:10.1021/ja028936q
- J. Org. Chem.1965, 308, 2635-2639, DOI: 10.1021/jo01019a030
- Org. Lett.1999, 14, 645-648, DOI: 10.1021/ol990723r
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者孙苏赟
概要
Mitsunobu反应可以将一级醇和二级醇的羟基转化成很多种其他的官能团,譬如胺、硫醚等,在有机合成中的应用十分广泛。

Mitsunobu反应中,三烷基膦是至关重要的试剂,可以将醇的羟基转化成为很好的离去基团,在当初此反应发明后,又有了很广泛的发展。对于亲核试剂,酸性要在pKa 10一下。当反应中加入ZnCl2或酰亚胺类化合物,反应物的构型可以实现完全的反转,若反应物是羧酸类化合物,构型的反转的酯化反应也是会发生的,这里可以将生成的酯再水解得到和反应物相反构型的羧酸化合物。此外,DIAD可以替换DEAD作为反应试剂,在反应中起到相同的作用。

反应机理[1]:

从机理上来看,PPh3先和DEAD作用,生成的中间体再和醇发生一系列质子转移过程,最后亲核试剂进攻醇的膦离子加合物,在此加合物中,具有一个很好的离去基团,亲核试剂进攻可以顺利的进行SN2,实现构型的反转,得到相反构型的化合物。
应用实例
(1) 酰胺化反应[2]:

(2) 实现不对称二级醇的构型反转[3]:

在这个反应中,先通过Mistunobu反应生成相反勾心的酯实现羟基构型的反转,再水解得到和反应物相反构型的二级醇。
(3) 叠氮取代[4]:

这个反应中使用了(PhO)2P(O)N3作为亲核试剂的来源:

(4) 实现大环的关环[5]:

除去之前所说传统的Mistunobu反应中使用的DIED和DEAD,还有一些改进的Mistubonu反应,使用的是改进的偶氮试剂和其他助剂:

例如:
(1) 烯丙基醇的反应[6]:

(2) CMBP的应用:

反应机理:

REFERENCES
- J. Org. Chem.1987, 52, 194, 235-4238 DOI:10.1021/jo00228a016
- J. Am. Chem. Soc. 1990, 112, 760, DOI: 10.1021/ja00158a040
- J. Am. Chem. Soc. 1999, 121, 5467, DOI: 10.1021/ja990404v
- J. Am. Chem. Soc.1999, 121, 33, 7702-7703,DOI: 10.1021/ja991714g
- J. Am. Chem. Soc. 2003, 125, 27, 8112-8113, DOI: 10.1021/ja036011k
- Trahedron Lett., 1994 35 5081 DOI: 1016/S0040-4039(00)73326-0
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
使用腈的苯胺衍生物的O-酰化反应。 该反应中,苯胺无需事先保护就行进行。
众所周知,对于苯胺的Friedel-Crafts酰基化,由于氮原子上的孤对电子对会与路易斯酸发生配位,导致芳香环的电子密度下降,使得反应很难进行。
而本反应通过使用两种路易斯酸,回避了上述问题。
通常的路易斯酸的组合是BCl3-AlCl3、而使用与氯原子亲和性更高的GaCl3,可以使得反应在更加温和的条件下进行。
基本文献
・Sugasawa, T.; Toyoda, T.; Adachi, M.; Sasakura, K. J. Am. Chem. Soc. 1978, 100, 4842. DOI: 10.1021/ja00483a034
・Douglas, A. W.; Abramson, N. L.; Houpis, I. N.; Karady, S.; Molina, A.; Xavier, L. C.; Yasuda, N. Tetrahedron Lett. 1994, 35, 6807. doi:10.1016/0040-4039(94)85010-0
・Prasad, K.; Lee, G. T.; Chaudhary, A.; Girgis, M. J.; Streemke, J. W.; Repic, O. Org. Proc. Res. Dev. 2003, 7, 723. DOI: 10.1021/op0340659
反应机理
参考:Tetrahedron Lett.1994, 35, 6807.

反应实例
使用菅沢反应进行的吲哚合成[1]

参考文献
[1] Pei, T.; Tellers, D. M.; Streckfuss, E. C.; Chen, C.-y.; Davies, I. W. Tetrahedron2009, 65, 3285. doi:10.1016/j.tet.2008.11.026
外部链接
・Lewis Acid Catlaysis – Wikipedia
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
ONSH(oxidative nucleophilic aromatic substitution of hydrogen) 反应是在氧化剂(如KMnO4, AgPy2MnO4, AgOAc, DDQ, CAN, Pb(OAc)4等,该反应所用的氧化剂必须与σH配合物及亲核试剂能够兼容)作用下通过σH配合物(σH adduct)进行的各类亲核试剂(如氮亲核试剂胺、硫亲核试剂及碳负离子等)对无离去基的缺电子芳烃或无离去基的芳香杂环化合物中的氢原子的亲核取代反应(有离去基存在时,则发生VNS(vicarious nucleophilic substitution)反应)。该反应在1981年由H. C. van der Plas首次报道[1]-[3]。目前,该反应已广泛应用于各类芳香化合物的构建[4]-[6]。
基本文献
- [1] A. Counotte-Potman, H. C. van der Plas, J. Heterocycl. Chem.1981, 18, 123. doi: 10.1002/jhet.5570180125.
- [2] H. C. van der Plas, Adv. Heterocycl. Chem. 2004, 86, 1. doi: 10.1016/S0065-2725(03)86001-4.
- [3] M. Makosza, K. Wojciechowski, Chem. Rev. 2004, 104, 2631. doi: 10.1021/cr020086+.
- [4] A. V. Gulevskaya, A. F. Pozharskii, Russ. Chem. Bull. 2008, 57, 913. doi: 10.1007/s11172-008-0127-3.
- [5] S. Verbeeck, W. A. Herrebout, A. V. Gulevskaya, J. Benjamin B. van der Veken, U. W. Maes, J. Org. Chem. 2010, 75, 5126. doi: 10.1021/jo100858n .
- [6] M. Makosza, Chem. Soc. Rev., 2010, 39, 2855. doi: 10.1039/B822559C.
反应机理

反应实例
芳基苯胺的合成[1]

硝基取代芳香氨基酸的合成[2]

嘧啶并哒嗪类化合物的杂环化 [3]

实验步骤
-78°C,氩气气氛下,向亲核试剂 (1 eq.) 的四氢呋喃溶液 (底物浓度为0.1 M)及硝基芳烃(2 eq.)的DMF (底物浓度为1 M) 溶液中滴加t-BuOK (1.3 eq.)的四氢呋喃溶液 (浓度为1.0 M)。维持-78°C,将上述反应混合物搅拌30 min后,加入氧化剂(1.2 eq.)的四氢呋喃溶液 (浓度为1.2 M)进行处理。5 min后,将上述混合物升至室温,加入稀盐酸及水进行处理。之后,采用DCM进行萃取,合并的有机相采用硅胶柱色谱(正己烷/乙酸乙酯体系作为洗脱剂)进行分离纯化,获得最终目标产物。
实验技巧
参考文献
- [1] V. V. Khutorianskyi, M. Sonawane, M. Pošta, B. Klepetářováa, P. Beier, Chem. Commun. 2016, 52, 7237. doi: 10.1039/C6CC02235A.
- [2] M. Mąkosza, M. Chromiński, D. Sulikowski, ARKIVOC 2011, 82. doi: HH-5827VP.
- [3] A. V. Gulevskaya, A. F. Pozharskii, Russ. Chem. Bull. 2008, 57, 913. doi: 10.1007/s11172-008-0127-3.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Nakata缩硫酮化-内酯化(Nakata thioketalization-lactonization) 是通过BF3·OEt2促进的缩酮与1,3-丙二硫醇之间的串联硫缩酮化/内酯化过程。该反应由日本理化研究院(The Institute of Physical and Chemical Research , Riken)的中田忠(Nakata Tadashi)研究组在1983年首次报道。目前该反应已成功应用于一些生物活性天然产物全合成的关键步骤。
基本文献
[1]T.Nakata, S.Takao, M.Fukui, T.Tanaka, T.Oishi, Tetrahedron Lett. 1983, 24, 3873.doi:10.1016/S0040-4039(00)94299-0.反应机理

反应实例
官能团化syn-1,3 ,5,7-四醇的合成[1]

Bryostatin 11底部片段(bottom half fragment)的合成[2]

实验步骤
将缩酮的(175 mg, 0.46 mmol) MeNO2-DCM溶液(2:1 v/v,底物浓度为0.15 M)冷却至-45°C,之后加入1,3-丙二硫醇(3.3eq.)与BF3·OEt2。将上述反应混合物逐渐升温至0°C(升温时间超过1h)。反应结束后,将上述反应液直接采用硅胶柱色谱进行分离纯化,获得相应目标产物。
实验技巧
参考文献
- [1]T.Nakata, S.Takao, M.Fukui, T.Tanaka, T.Oishi, Tetrahedron Lett. 1983, 24, 3873.doi:10.1016/S0040-4039(00)94299-0.
- [2] K. Nakagawa-Goto, M. T. Crimmins, Synlett 2011, 11, 1555.xdoi:10.1055/s-0030-1260784.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Natsume 吲哚合成(Natsume indole synthesis)是通过采用Lewis酸 (如AlCl3及 BF3·Et2O)参与的吡咯环的亲电取代反应,随后,经官能团化过程而进行的吲哚环的合成。该反应由日本Research Foundation Itsuu Laboratory的夏目充隆(Natsume Mitsutaka)研究室1990年首次报道。目前,该反应已经广泛应用于各类烷基取代吲哚类化合物的合成及吲哚类生物碱的全合成。
基本文献
[1] H. Muratake, M. Natsume, Heterocycles 1990, 31, 683. doi: 10.3987/COM-90-5333. [2] K. Okabe, H. Muratake; M. Natsume, Tetrahedron 1990, 46, 5113. doi: 10.1016/S0040-4020(01)87818-2.反应机理

反应实例
4-烷基吲哚的合成[1]

吲哚类生物碱pendolmycin的全合成[2]

实验步骤
- Friedel-Crafts酰基化[3] [4]
25oC下,将BF3•OEt2(2.2 eq.)加入到乙酸酐(1.1 eq.)的DCE溶液(底物浓度为0.36 M)中。将上述反应混合物搅拌10 min后,加入吡咯(1eq.)的DCE溶液(底物浓度为1M),将上述混合物在该温度下搅拌,直至反应结束。反应结束后,加入冷水淬灭反应,并用DCM进行萃取。将合并的有机相减压除去溶剂后获得粗产物。将粗产物采用硅胶柱色谱(乙酸乙酯/甲苯 1:4 v/v作为洗脱剂)进行分离纯化,获得相应乙酰基化产物。最后,在正己烷溶剂中进行重结晶获得纯净的酰基化产物。
- Grignard 反应
-20°C,氩气气氛下,将2-(1,3-二噁烷-2-基)乙基溴化镁(1eq.)的THF (浓度为溶液加入到酰基吡咯(1eq.)的THF (底物浓度为0.09 M)溶液中。将上述反应混合物搅拌15 min后,加入sat. NH4Cl/H2O淬灭反应,淬灭结束后,再加入DCM进行萃取。将合并的有机相采用无水硫酸钠进行干燥后,减压除去溶剂。粗产物采用Prep-TLC(正己烷/EtOAc 5:2 v/v作为展开剂)分离纯化获得相应成环前体。
- 吲哚成环反应
将第二步反应产生的成环前体(1eq.)的6% H2SO4-异丙醇溶液(底物浓度为0.03 M)加热回流30min。反应结束后,将反应液进行冷却后加水稀释,再加入DCM进行萃取。将合并的有机相采用无水硫酸钠进行干燥后,减压除去溶剂。粗产物采用Prep-TLC(正己烷/EtOAc 9:l v/v作为展开剂)分离纯化获得最终目标产物。
实验技巧
参考文献
- [1] H. Muratake, M. Natsume, Heterocycles 1990, 31, 683. doi: 10.3987/COM-90-5333.
- [2] K. Okabe, H. Muratake; M. Natsume, Tetrahedron 1990, 46, 5113. doi: 10.1016/S0040-4020(01)87818-2.
- [3] J. Rokach, P. Hamel, M. Kakushima, G. M. Smith, Tetrahedron Lett. 1981, 22, 4901. doi: 10.1016/S0040-4039(01)92377-9.
- [4] M. Kakushima, P. Hamel, R. Frenette, I. Rokach, J. Org. Chem. 1983,48, 3214. doi: 10.1021/jo00167a014.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
芳香族化合物通常对亲核取代反应是惰性的、然而对于具有强吸电子取代基如氰基或硝基的芳环、或者反应经由重氮,苯炔等特殊活性基团的情况的时候,也能发生亲核取代反应。
基本文献
- SN(ANRORC): Henk C. Van der Plas, Acc. Chem. Res. 1978, 11, 462. doi:10.1021/ar50132a005
- Vicarious nucleophilic substitution: Makosza, M.; Winiarski, J. Acc. Chem. Res.1987, 20, 282. doi:10.1021/ar00140a003
反应机理
机制因底物而异。 粗略可以分为以下几类。
①SNAr机理(加成-消除):吸电子基团的存在是必要条件。卤素通常用作离去基团。反应活性与正常SN1 / SN2反应呈相反的趋势、也就是F>Cl>Br>I
。在亲核加成之后与阴离子离域的中间体被称为Meisenheimer Complex。

②SN1机理(消除-加成型):仅限于含有重氮盐基团这样的强力离去基团存在的情况,反应通过苯鎓阳离子中间体进行。

③苯炔机理: 经常发生在使用具有强碱性的强亲核剂的场合。

④SN(ANRORC)机理:经过一次开环过程的特殊的机理。

⑤Vicarious Nucleophilic Substitution:
在离去基团存在于亲核侧而不是芳环的特殊情况下。从形式上而言,芳香环上的氢被置换。

⑥SRN1机理(Free Radical): 当硫醇盐或者碘等作为离去基团的场合,也就是具有氧化还原特性的基团的底物。
反应实例
催化芳香族的亲核取代反应[1]

实验步骤
在反应容器中加入、Ru(cod)(2-methylallyl)2 (6.4 mg, 0.020 mmol),1,5-bis(diphenylphosphino)pentane (12.3 mg, 0.028mmol), TfOH (3.5 μL, 0.040 mmol) 与1,4-dioxane(2 mL)。然后在这个悬浊液中继续加入 p-fluorotoluene (2.0 mmol), morpholine (0.40 mmol), triethylamine (0.40 mmol) 与triethylsilane (0.40 mmol)、加热回流反应24小时。溶剂减压旋蒸除去后,用柱色谱纯化(TLC:hexane/AcOEt =10/1)得到72%产率的产物 [1]
实验技巧
参考文献
[1] Otsuka, M.; Endo, K.; Shibata, T. Chem. Commun. 2010, 46, 336. doi:10.1039/b919413d
相关书籍(日亚价格)

Modern Nucleophilic Aromatic Substitution
- 著者Francois Terrier
- 参考価格¥ 23,336
価格¥ 17,770(2017/12/27 15:28時点) - 出版日2013/08/05
- 商品ランキング89,181位
- ハードカバー488ページ
- ISBN-103527318615
- ISBN-139783527318612
- 出版社Wiley-VCH
相关链接
- Nucleophilic Aromatic Subsitituion – WikipediaANRORC Mechanism– Wikipedia
- Vicarious nucleophilic substitution – Wikipedia
- MeisenheimerComplex – Wikipedia
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Kita酯化(Kita esterification)是通过形成1-乙氧乙烯基酯(EVE)进行的羧酸与醇的酯化过程。该方法在1986年由日本立命馆大学(Ritsumeikan University)的北泰行(Yasuyuki Kita )研究组首次报道。实验中1-乙氧乙烯基酯通常由羧酸与乙氧基乙炔在催化量的[RuCl2(p-cymene)]2 (最初采用Hg(II)化合物作为催化剂)作用下原位制备。另外,该课题组研究发现上述条件同样适用于N-酰基化合物(酰胺)的合成及各类酸敏与碱敏底物的大环内酯化,具有优良的底物适用性与官能团兼容性。
基本文献
- [1]Y. Kita, S. Akai, N. Ajimura, M. Yoshigi, T. Tsugoshi, H. Yasuda, Y. Tamura, J. Org. Chem., 1986, 51, 4150. doi: 10.1021/jo00372a010.
- [2] Y. Ohba, M. Takatsuji, K. Nakahara, H. Fujioka, Y. Kita, Chem. Eur. J., 2009, 15, 3526. doi: 10.1002/chem.200801548.
反应机理

反应实例
七元环内酰胺的合成[1]

大环内酯的合成[2]

水溶性的oxaunomycin衍生物的合成[3]

实验步骤
EVE的原位合成:氮气气氛下,将[RuCl2 (p-cymene)]2 (0.02 eq.) 与乙氧基乙炔(2 eq.)缓慢加入到羟基酸的(1 eq.)丙酮(0.05 M)溶液中。将上述反应液在室温下搅拌 6 h。反应结束后,将上述反应液进行真空浓缩。所得粗产物采用硅胶柱色谱分离纯化(hexane/EtOAc 8:1) 获得相应EVE酯。
Kita酯化: 将所得EVE酯用 DCE进行稀释(0.001 M),并将其用注射泵缓慢加入到高度稀释的 pTsOH (0.05m in DCE/CH3CN 1:1, 0.05M) 的DCE (0.0003 M) 溶液中(超过10 h后,滴加结束,并维持反应液温度为 80 oC)。将上述反应混合物在80 oC下继续搅拌1 h,并将反应液冷却至室温。再向上述反应液中加入Et3N (ca. 1.2 eq.),并将其进行减压浓缩。所得粗产物采用硅胶柱色谱进行分离纯化 (hexane/Et2O 20:1) 获得最终目标产物。
实验技巧
参考文献
- [1] Y. Ohba, M. Takatsuji, K. Nakahara, H. Fujioka, Y. Kita, Chem. Eur. J., 2009, 15, 3526. doi: 10.1002/chem.200801548.
- [2] B. M. Trost, J. D. Chisholm, Org. Lett. 2002, 4, 3743. doi:10.1021/ol026726c.
- [3] Y. Kita, H. Maeda, K. Omori, T. Okuno, Y. Tamura, Synlett, 1993, 273. doi: 10.1055/s-1993-22428.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!