概要
Balci-Güven重排 (Balci-Güven rearrangement)是在均相金催化剂存在下,N-炔丙基取代吲哚甲醛肟或N-炔丙基取代吡咯甲醛肟通过重排过程,生成N-氧化物或酮肟的反应[1]。该反应由土耳其Giresun大学化学系 (Department of Chemistry, Giresun University)的M. Balci与中东技术大学化学系 (Department of Chemistry, Middle East Technical University) 的S. Güven在2015年共同报道[1]。

该反应条件温和,目标产物产率良好,在各类N-氧化物与酮肟分子的合成中具有广阔的应用前景。
基本文献
- [1] S. Güven, M. S. Ozer, S. Kaya, N. Menges, M. Balci, Org. Lett. 2015, 17, 2660. doi: 10.1021/acs.orglett.5b01041.
反应机理
1. 生成N-氧化物

2. 生成酮肟

参考文献
- [1] S. Basceken, Struct. Chem. 2020, ASAP. doi: 10.1007/s11224-020-01531-x.
反应实例
N-氧化物的合成[1]

酮肟的合成[1]

实验步骤
1. 生成N-氧化物

2. 生成酮肟

参考文献
- [1] S. Güven, M. S. Ozer, S. Kaya, N. Menges, M. Balci, Org. Lett. 2015, 17, 2660. doi: 10.1021/acs.orglett.5b01041.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
1961年开始,挪威Oslo大学( University of Oslo)的Skattebøl研究组报道了偕二溴代环丙烷在有机锂试剂作用下,转化为联烯的反应[1]-[4]。

之后,该课题组在研究乙烯基偕二溴代环丙烷在该条件下的反应时,发现形成的并非联烯,而是形成环戊二烯类化合物[4]-[6]。

上述反应称为Skattebøl重排(Skattebøl rearrangement)。目前,该反应已经广泛应用于各类联烯与环戊二烯化合物的合成及各类特殊结构的有机分子的构建[7]-[10]。
基本文献
- [1] L. Skattebøl, Tetrahedron Lett. 1961, 167. doi: 10.1016/S0040-4039(01)84059-4.
- [2] L. Skattebøl, Acta. Chem. Scand. 1963, 17, 1683. doi: 10.3891/acta.chem.scand.17-1683.
- [3] L. Skattebøl. J. Org. Chem. 1966, 31, 2789. doi: 10.1021/jo01347a014.
- [4] L. Skattebøl, Tetrahedron 1967, 23, 1107. doi: 10.1016/0040-4020(67)85060-9.
- [5] K. H. Holm, L. Skattebøl, J. Am. Chem. Soc. 1977, 99, 5480. doi: 10.1021/ja00458a042.
- [6] K. H. Holm, L. Skattebøl, Tetrahedron Lett. 1977, 2347. doi: 10.1016/S0040-4039(01)83760-6.
- [7] S. C. Sutton, M. H. Nantz, S. R. Parkin, Organometallics 1993, 12, 2248. doi: 10.1021/om00030a040.
- [8] L. K. Sydnes, Chem. Rev. 2003, 103, 1133. doi: 10.1021/cr0100087.
- [9] L. A. Paquette, K. E. Green, R. Gleiter, W. Schafer, J. C. Gallucci, J. Am. Chem. Soc. 1984, 106, 8232. doi: 10.1021/ja00338a037.
- [10] P. Charumilind, L. A. Paquette, J. Am. Chem. Soc. 1984, 106, 8225. 10.1021/ja00338a036.
反应机理

- [1] W. R. Moore, H. R. Ward, J. Org. Chem. 1962, 27, 4179. doi: 10.1021/jo01059a013.
- [2] U. H. Brinker, J. Ritzer, J. Am. Chem. Soc. 1981, 103, 2116. doi: 10.1021/ja00398a052.
- [3] A. Azizoglu, M. Balci, J.-L. Mieusset, H. U. Brinker, J. Org. Chem. 2008, 73, 8182. doi: 10.1021/jo8011144.
- [4] L. A. Paquette, R. Taylor, J. Am. Chem. Soc. 1977, 99, 5708. doi: 10.1021/ja00459a031.
反应实例
环戊二烯类化合物的合成[1]-[3]

ansa-二茂钛的合成[4]

联烯类化合物的合成[5]

实验步骤
联烯合成
将1,1-二溴环丙烷 (1 eq.)用无水乙醚进行稀释(维持底物浓度为4 M)后,置于干冰/丙酮浴中冷却,维持温度在-30到-40oC。将甲基锂的乙醚溶液 (1.5 M, 4eq.)滴加到上述均相混合物中,搅拌30min。将上述反应混合物继续搅拌30min后,加入水进行淬灭。淬灭完成后,分出乙醚相,水相继续采用少量乙醚进行萃取。合并有机相,用水洗涤至中性后,加入无水硫酸钠进行干燥。随后,采用Dixon环填料柱 (Dixon ring-packed column)进行分馏获得相应联烯产物。
环戊二烯类化合物的合成
向火焰干燥的圆底烧瓶中加入偕二溴环丙烷 (1 eq.),之后加入无水乙醚进行稀释 (维持底物浓度为4 M)。将上述溶液置于冰水浴中搅拌冷却。随后,将甲基锂的乙醚溶液 (1.5 M, 4eq.)滴加到上述均相混合物中,移除冰水浴,继续搅拌直至反应结束。反应结束后,加入冰水进行淬灭。淬灭完成后,分出乙醚相,水相继续采用少量乙醚进行萃取。将合并的有机相用水洗涤至中性后,加入无水硫酸钠进行干燥,减压除去溶剂。将残余物加入等体积正己烷进行稀释,通过中性氧化铝短柱进行过滤。小心除去溶剂后,在90°C下,对残余物进行Kugelrohr蒸馏,获得相应环戊二烯产物。
参考文献
- [1] L. A. Paquette, M. L. McLaughlin, Org. Synth. 1990, 68, 220. doi: 10.15227/orgsyn.068.0220.
- [2] McLaughlin, M. L.; McKinney, J. A.; Paquette, L. A. Tetrahedron Lett. 1986, 27, 5595. doi: 10.1016/S0040-4039(00)85274-0.
- [3] Paquette, L. A.; Gugelchuk, M.; McLaughlin, M. L. J. Org. Chem. 1987, 52, 4732. doi: 10.1021/jo00230a015.
- [4] S. C. Sutton, M. H. Nantz, S. R. Parkin, Organometallics 1993, 12, 2248. doi: 10.1021/om00030a040.
- [5] L. Skattebøl. J. Org. Chem. 1966, 31, 2789. doi: 10.1021/jo01347a014.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Chen-Mapp重排 (Chen-Mapp rearrangement)又称为[3,3]-phosphorimidate重排或[3,3]-氮杂-磷杂-氧杂-Cope重排 ([3,3]-aza-phospha-oxa-Cope rearrangement)[1],是allylic phosphorimidate或propargylic phosphorimidate (通常由烯丙醇或炔丙醇与有机膦或亚磷酸酯及有机叠氮化合物原位反应生成[2]-[3])在加热[1]或过渡金属钯催化[4]-[7]条件下进行的分子内[3,3]-σ-重排过程,生成phosphoramidate的反应。该反应在2004年由美国Michigan大学化学与医药化学系 (Departments of Chemistry and Medicinal Chemistry, University of Michigan)的B. Chen与A. K. Mapp首次报道[1]。

2004年,R. A. Batey采用钯催化剂参与该重排,使反应可以在较为温和的条件下进行,并获得良好产率[5]。

2004年,R. A. Batey研究组采用手性N, N′-二甲基环己二胺辅基,成功实现反应的非对映选择性控制[6]。

同时,R. A. Batey采用手性催化剂COP(chiral cobalt oxazoline palladacycles)-Cl,成功实现N-保护型烯丙胺的对映选择性合成[6]。

2010年,A. K. Mapp将底物扩展至propargylic phosphorimidate,成功完成了联烯基酰胺 (allenamide)的合成[7]。

目前,该反应广泛应用于各种N-保护型烯丙胺 (N-protected allylic amines)与联烯基酰胺类化合物的合成,而N-保护型烯丙胺、烯丙胺与联烯基酰胺类化合物是有机合成方法学研究及天然产物全合成的关键砌块[8]-[19]。
基本文献
- [1] Chen, A. K. Mapp, J. Am. Chem. Soc. 2004, 126, 5364. doi: 10.1021/ja0490469.
- [2] G. Gololobov, I. N. Zhmurova, L. F. Kasukhin, Tetrahedron 1981, 37, 437. doi: 10.1016/S0040-4020(01)92417-2.
- [3] G. Gololobov, L. F. Kasukhin, Tetrahedron 1992, 48, 1353. doi: 10.1016/S0040-4020(01)92229-X.
- [4] Chen, A. K. Mapp, J. Am. Chem. Soc.2005, 127, 6712. doi: 10.1021/ja050759g.
- [5] E. Lee, R. A. Batey, Angew. Chem. Int. Ed. 2004, 43, 1865. doi: 10.1002/anie.200353284.
- [6] E. Lee, R. A. Batey, J. Am. Chem. Soc. 2005, 127, 14887. doi: 10.1021/ja054161k.
- [7] M. Danowitz, C. E. Taylor, T. M. Shrikian, A. K. Mapp, Org. Lett. 2010, 12, 2574. doi: 10.1021/ol1007845.
- [8] Cativiela, M. D. Diaz-de-Villegas, Tetrahedron: Asymmetry 1998, 9, 3517. doi: 10.1016/S0957-4166(98)00391-7.
- [9] Hayashi, A. Yamamoto, Y. Ito, E. Nishioka, H. Miura, K. Yanagi, J. Am. Chem. Soc. 1989, 111, 6301. doi: 10.1021/ja00198a048.
- [10] M. Trost, D. L. Vanvranken, J. Am. Chem. Soc. 1993, 115, 444. doi: 10.1021/ja00055a013.
- [11] Airiau, N. Girard, M. Pizzeti, J. Salvadori, M. Taddei, A. Mann, J. Org. Chem. 2010, 75, 8670. doi: 10.1021/jo101776y.
- [12] Burgess, M. J. Ohlmeyer, J. Org.Chem. 1991, 56, 1027. doi: 10.1021/jo00003a024.
- [13] J. T. Hyland, L. S. Hegedus, J. Org. Chem. 2006, 71, 8658. doi: 10.1021/jo061340r.
- [14] R. Berry, R. P. Hsung, J. E. Antoline, M. E. Petersen, R. Challeppan, J. A. Nielson, J. Org. Chem. 2005, 70, 4038. doi: 10.1021/jo0503411.
- [15] L. Wei, R. P. Hsung, H. Xiong, J. A. Mulder, N. T. Nkansah, Org. Lett. 1999, 1, 2145. doi: 10.1021/ol990345q.
- [16] Hayashi, R. P. Hsung, J. B. Feltenberger, A. G. Lohse, Org. Lett. 2009, 11, 2125. doi: 10.1021/ol900647s.
- [17] Lu, R. Hayashi, R. P. Hsung, K. A. DeKorver, A. G. Lohse, Z. Song, Y. Tang, Org. Biomol. Chem. 2009, 7, 3331. doi: 10.1039/B908205K.
- [18] Ramage, D. Hopton, M. J. Parrott, G. W. Kenner, G. A. Moore, J. Chem. Soc., Perkin Trans. 1 1984, 1357. doi: 10.1039/P19840001357.
- [19] F. Zhao, S. K. Xi, A. T. Song, G. J. Ji, J. Org. Chem. 1984, 49, 4549. doi: 10.1021/jo00197a050.
反应机理
Chen-Mapp重排反应的驱动力为P=O双键比P=N双键具有更高的BDE。
allylic phosphorimidate底物的Chen-Mapp重排[1]-[3]

过渡金属催化的allylic phosphorimidate底物的Chen-Mapp重排[4]

手性辅基参与的Chen-Mapp重排[5]

COP-X参与的Chen-Mapp重排[6]-[7]

propargylic phosphorimidate底物的Chen-Mapp重排

参考文献
- [1] T. A. Albright, W. J. Freeman, E. E. Schweizer, J. Org. Chem. 1976, 41, 2716. doi: 10.1021/jo00878a013.
- [2] J. Barluenga, F. Palacios, Org. Prep. Proced. Int. 1991, 23, 1. doi: 10.1080/00304949109458286.
- [3] A. Alexakis, S. Mutti, P. Mangeney, J. Org. Chem. 1992, 57, 1224. doi: 10.1021/jo00030a034.
- [4] E. E. Lee, R. A. Batey, Angew. Chem. Int. Ed. Engl. 2004, 43, 1865. doi: 10.1002/anie.200353284.
- [5] E. E. Lee, R. A. Batey, J. Am. Chem. Soc. 2005, 127, 14887. doi: 10.1021/ja054161k.
- [6] D. J. Cassar, H. Roghzai, D. Villemin, P. N. Horton, S. J. Coles, J. C. Richards, Organometallics 2015, 34, 2953. doi: 10.1021/acs.organomet.5b00282.
- [7] J. S. Cannon, L. E. Overman, Acc. Chem. Res. 2016, 49, 2220. doi: 10.1021/acs.accounts.6b00398.
反应实例
烯丙胺的合成[1] [2]

联烯基酰胺类化合物的合成[3]

实验步骤
allylic phosphorimidate底物的Chen-Mapp重排
向冰水浴冷却的乙醚中加入烯丙醇 (1.25 eq.)Et3N (1.25 eq.), 随后加入2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane或氯亚磷酸二乙酯的乙醚溶液 (浓度为1M,1.25 eq.)。将上述溶液搅拌20 min,随后采用标准Schlenk技术进行过滤,除去生成的Et3N•HCl沉淀。通过真空除去乙醚,后将残余物溶于适量二甲苯。向上述二甲苯溶液中滴加冰水浴冷却的苄基叠氮的二甲苯溶液 (浓度为2M,1 eq.)。注意:在使用烯丙基叠氮参与本反应时,因其具有挥发性,因此需要过量加入 (通常为3 eq.)。将上述反应混合物缓慢升至室温 (升温时间高于1h),后将上述混合物在80 °C下加热1 h, 随后加入适量二甲苯进行稀释,继续加入回流4 h。4 h后,将上述反应液冷却,采用快速柱色谱直接进行纯化 (正己烷/EtOAc 1:9 v/v作为洗脱剂),获得相应phosphoramidate产物。
过渡金属催化的Chen-Mapp重排
方法 A. tosyl iminodiazaphospholidines在室温条件下进行的重排
N2气氛下,向火焰干燥过的圆底烧瓶中加入iminodiazaphospholidine (1 eq.) 的CH2Cl2溶液(浓度为0.12 M),随后加入二氯双(乙腈)合钯(II) (0.05 eq.)。将上述反应混合物在N2气氛下进行搅拌,并通过31P-NMR进行监控,直至反应结束。反应结束后,将反应液在真空条件下进行浓缩,随后将其溶于少量二氯甲烷,并通过硅胶柱色谱进行分离纯化 (EtOAc/正己烷 2:1 v/v作为洗脱剂),获得相应重排产物。
方法 B. tosyl iminodiazaphospholidines在加入条件下进行的重排
N2气氛下,向火焰干燥过的圆底烧瓶中装入回流冷凝管,随后加入iminodiazaphospholidine (1 eq.) 的CH2Cl2溶液 (浓度为0.12 M),随后加入二氯双(乙腈)合钯(II) (0.05 eq.)及适量新活化的4Å分子筛。继续维持在N2气氛下,将上述反应混合物45°C下加热搅拌,并通过31P-NMR进行监控,直至反应结束。反应结束后,将反应液冷却至室温,并在真空条件下进行浓缩,随后将其溶于少量二氯甲烷,并通过硅胶柱色谱进行分离纯化 (EtOAc/正己烷 2:1 v/v作为洗脱剂),获得相应重排产物。
方法 C. tosyl iminodiazaphospholidines的不对称重排
N2气氛下,向火焰干燥过的尖底反应瓶 (conical vial)中,加入甲苯 (维持底物浓度为1 M)、iminodiazaphospholidine (1 eq.)、 COP.Cl (0.05 eq)及AgTFA (0. 1 eq.). 将反应瓶密封,并在45°C下加热搅拌,并通过31P-NMR进行监控,直至反应结束。反应结束后,将反应液冷却至室温,并在真空条件下进行浓缩。随后,将其溶于少量二氯甲烷,并通过硅胶柱色谱进行分离纯化 (EtOAc/正己烷 2:1 v/v作为洗脱剂),获得相应重排产物。
propargylic phosphorimidate底物的Chen-Mapp重排
向冰水浴冷却的乙醚中加入炔丙醇 (1.3 eq.)与Et3N (1.3 eq.), 随后加入氯亚磷酸二乙酯 (1.3 eq.)。将上述溶液搅拌20 min,随后进行过滤,除去生成的Et3N•HCl沉淀。在真空条件下除去乙醚,后将残余物溶于适量二氯甲烷。室温条件下,向上述反应混合物中滴加苄氧羰基叠氮 (1 eq.)。将上述反应混合物继续搅拌2h,真空条件下除去DCM, 将粗产物采用硅胶柱色谱分离纯化(首先用正己烷/EtOAc 1:5v/v洗脱剂, 随后更换正己烷/EtOAc 1:2.5 v/v洗脱剂,进行梯度洗脱),获得相应phosphorimidate。注意:phosphorimidate 放置时易分解,需要立即进行分离纯化。
向phosphorimidate (1.0 eq)的DCM溶液 (浓度为0.1 M)中加入PdCl2(CH3CN)2 (0.03 eq)。将上述混合物在室温下进行搅拌, 并通过TLC进行监控 (正己烷/EtOAc 1:1 v/v作为展开剂),直至反应结束(5-7 h)。反应结束后,在真空条件下除去二氯甲烷,并通过硅胶柱色谱进行分离纯化 (首先用正己烷/EtOAc 1:5v/v洗脱剂, 随后更换正己烷/EtOAc 1:2.5 v/v洗脱剂,进行梯度洗脱),获得相应联烯酰胺重排产物。注意:延长反应时间会出现产物分解与差向异构化。
实验安全须知
无机与有机叠氮化物具有较强的毒性与潜在的爆炸性,需要在通风极其优良的通风橱内小心仔细进行操作。并穿戴SSG防化手套。
参考文献
- [1] B. Chen, A. K. Mapp, J. Am. Chem. Soc. 2004, 126, 5364. doi: 10.1021/ja0490469.
- [2] A. M. Danowitz, C. E. Taylor, T. M. Shrikian, A. K. Mapp, Org. Lett. 2010, 12, 2574. doi: 10.1021/ol1007845.
- [3] A. M. Danowitz, C. E. Taylor, T. M. Shrikian, A. K. Mapp, Org. Lett. 2010, 12, 2574. doi: 10.1021/ol1007845.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者:alberto-caeiro
Doyle–Kirmse Reaction是重氮化合物与烯丙基硫化物,烯丙基碘发生反应,得到[2,3]-Sigmatropic重排产物。最早由W. Kirmse于1968年报道[1],1981年M. P. Doyle对其进行改进[2]。
反应机理如下:重氮化合物在金属作用下生成金属卡宾复合物,随后受到杂原子的进攻,形成杂原子叶立德(Free or Metal Bound)[3],随后发生[2,3]-Sigmatropic Rearrangement得到产物。各种重氮化合物都能反应,烯丙基碘化物和烯丙基硫化物一般是亲核试剂,催化的过渡金属一般为铜或者铑。

应用实例
四川大学的冯小明院士通过使用他们发展的手性N-O配体,实现了不对称的Doyle-Kirmse反应,其中吡唑酰胺对手性的控制有着重要的作用[4]。北京大学的王剑波教授则通过铑和铜催化的不对称的Doyle-Kirmse反应,实现了手性三氟甲硫基的构建[5]。

美国西南医学中心的Uttam Tambar教授实现了铜催化不对称烯丙基碘化物的Doyle-Kirmse反应,其中他们发现,通过配体的调控,可实现[1,2],[2,3]- Sigmatropic Rearrangement反应的选择[6]。

参考文献
- Kirmse, W. Kapps, M. Reaktionen des Diazomethans mit Diallylsulfid und Allyläthern unter Kupfersalz-Katalyse. Chemische Berichte, 1968, 101: 994. doi:10.1002/cber.19681010333;
- Michael P. Doyle, William H. Tamblyn, Vahid Bagheri J. Org. Chem., 1981, 46 , 5094–5102 doi:10.1021/jo00338a008;
- Katharina J. Hock, Rene M. Koenigs, Angew. Chem., Int. Ed., 2017, 56, 13566, doi.org/10.1002/anie.201707092;
- Xiaobin Lin, Yu TangWei Yang, Fei TanLili Lin, Xiaohua Liu, Xiaoming Feng, J. Am. Chem. Soc., 2018, 140, 3299, doi.org/10.1021/jacs.7b12486;
- Zhikun Zhang, Zhe Sheng, Weizhi Yu, Guojiao Wu, Rui Zhang, Wen-Dao Chu, Yan Zhang, Jianbo Wang, Nat. Chem., 2017, 9, 970. doi.org/10.1038/nchem.2789;
- a. Bin Xu, Uttam K. Tambar. J. Am. Chem. Soc., 2016, 138, 12073. DOI: 10.1021/jacs.6b08624; b. Bin Xu, Uttam K. Tambar. Angew. Chem., Int. Ed., 2017, 56, 9868. DOI: 10.1002/anie.201705317.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
背景
酚类化合物是一类重要的有机合成前体,它广泛存在于自然界和生物体系。传统的制备酚类化合物的方法包括卤代苯水解法、异丙苯氧化法、重氮盐水解法等,此外,双烯酮-酚重排反应也被广泛用于酚类化合物的制备。早在1893年,化学家A. Andreocci就发现蛔虫药“山道年”(Santonin)在酸性条件下会生成另一种物质,但直到1930年“山道年”及其酸性条件下生成的产物结构才被确定。1946年,美国化学家A. L. Wilds和C. Djerassi首先将该反应用于Chrysene衍生物的合成并首次将其命名为Dienone-Phenol Rearrangement。[1]

“山道年”在酸性条件下的重排
反应概况
环己二烯酮化合物在酸或光化学诱导下,烃基发生分子内迁移生成多取代苯酚的反应被定义为“双烯酮-酚重排”。邻位或对位取代的环己二烯酮本身不具备芳香性,但产物苯酚具有较为稳定的芳香结构,因此该过程的巨大驱动力是“芳构化”,反应往往需要放出大量的热。通常情况下,该反应仅仅需要中等强度的酸介质,例如H2SO4/HOAc,Ac2O,Lewis酸等就可以获得可观产率,因而实际应用非常广泛。[2]

“双烯酮-酚重排”的反应通式
反应机理
“双烯酮-酚重排”反应的机理并不复杂,由于最常用的催化条件是酸,因此反应通常被认为涉及碳正离子中间体。首先环己二烯酮的羰基氧原子被质子化,氧鎓离子共振成相应的碳正离子后发生一系列重排,最后失去质子恢复稳定的芳香结构。根据反应机理我们不难推测不同基团发生迁移的难易程度,显然迁移后形成的碳正离子越稳定该基团也越容易迁移。一般而言,基团迁移大致为OR > Ph > Me,vinyl > Me。[3]

“双烯酮-酚重排”的反应机理
合成应用
- 多取代苯酚的制备[4]
2001年,A.G. Schultz组报道了一个由苯酚制备多取代苯酚的简洁方法。首先,通过Birch reduction-alkylation反应将叠氮烷基引入底物,TBHP进一步氧化后可构建环己二烯酮中间体。惰性非极性溶剂苯中,利用波长为366 nm的辐射光照射中间体,可以高区域选择性和高产率获得2,3,4-多取代苯酚。

多取代苯酚的制备
- 螺环吲哚酮在Lewis酸介导下的开环-关环重排[5]
2017年,天津大学赵康教授和杜云飞教授合作报道了螺环吲哚酮在非金属Lewis酸BF3Et2O催化下发生分子内迁移,高效合成了具有潜在生物活性的8-hydroxy-phenanthridin-6(5H)-one衍生物,初步的机理研究表明,反应可能涉及酰胺氮正离子中间体。

螺环吲哚酮在Lewis酸介导下的开环-关环重排
- Krempene B的全合成[6]
Krempene B是一类重要的甾体药物中间体,广泛用于甾体类药物、黄体酮类药物及生化研究。2012年,广西民族大学申利群教授以酰基保护的孕烯醇酮为起始原料,历经11步以较高产率实现了Krempene B的合成,其中的关键步骤是构建环己二烯酮中间体并利用“双烯酮-酚”重排构建苯酚骨架,最终他们以85%的高产率实现了这一转化。

“双烯酮-酚重排”在Krempene B合成中的应用
参考文献
- [1] Wilds A L, Djerassi C. J. Am. Chem. Soc. 1946, 68, 1715-1719. DOI: 10.1021/ja01213a010
- [2] Kurti L, Czakó B. Strategic applications of named reactions in organic synthesis[M]. Elsevier, 2005. pp. 142-143. ISBN: 0-12-429785-4
- [3] Wang H. Comprehensive Organic Name Reactions[M]. Wiley, 2010. pp. 900-904.
- DOI: 10.1002/9780470638859.conrr193
- [4] Guo Z, Schultz A G. Org. Lett. 2001, 3, 1177-1180. DOI: 10.1021/ol015637h
- [5] Guo X, Xing Q, Lei K, et al. Adv. Synth. Catal, 2017, 359, 4393-4398.
- DOI: 10.1002/adsc.201700728
- [6] Shen L-Q, Huang S-Y, Tang Y, et al. Steroids, 2012, 77, 1398-1402.
- DOI: 10.1016/j.steroids.2012.08.006
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Kuwajima-Reich重排(Kuwajima-Reichrearrangent)是在强碱(如烷基锂)诱导下,1-三甲基硅基炔丙醇发生重排,生成硅氧基联烯的反应。该反应在1980年由日本东京工业大学(Tokyo Institute of Technology)的桑岛功(KuwajimaIsao)研究组与美国Wisconsin大学(University of Wisconsin)的H. J.Reich研究组独立报道[1]-[4]。2011年,日本广岛大学(Hiroshima University)的武田敬(Takeda Kei)研究组报道了首例对映选择性Kuwajima-Reich重排。
 目前,该反应已经成为硅氧基联烯合成时最为常用的方法[5]。同时,采用不同的亲电底物将重排产生的联烯基锂捕获,可以方便地构建采用常规方法难以合成的各类有机分子[6]-[8]。
目前,该反应已经成为硅氧基联烯合成时最为常用的方法[5]。同时,采用不同的亲电底物将重排产生的联烯基锂捕获,可以方便地构建采用常规方法难以合成的各类有机分子[6]-[8]。


基本文献
- [1]I.Kuwajima, M. Kato, TetrahedronLett.1980, 21, 623. doi:10.1016/S0040-4039(01)85575-1.
- [2] H. J.Reich, R. E.Olson, M. C.Clark, J. Am. Chem. Soc. 1980, 102, 1423.doi:10.1021/ja00524a036.
- [3]I. Kuwajima, J. Organomet. Chem. 1985, 285, 137.doi:10.1016/0022-328X(85)87363-0.
- [4] H. J.Reich, E. K.Eisenhart, R. E.Olson,M. J. Kelly, J. Am. Chem. Soc.1986, 108, 7791. doi:10.1021/ja00284a051.
- [5] M.Sasaki, Y.Kondo, M. Kawahata, K.Yamaguchi, K.Takeda, Angew.Chem. Int. Ed. 2011, 123, 6499. doi:10.1002/ange.201102430.
- [6] R.Matsuoka, Y. Horiguchi, I. Kuwajima, Tetrahedron Lett. 1987, 28, 1299.doi:10.1016/S0040-4039(00)95352-8.
- [7] G.R. Boyce, S. Liu, J. S. Johnson, Org.Lett. 2012, 14, 652.doi:10.1021/ol2033527.
- [8]T. E. Reynolds, K. A. Scheidt, Angew.Chem. Int. Ed. Engl. 2007, 46, 7806.doi: 10.1002/anie.200702818.
反应机理

反应实例
- 酮类化合物的合成[1]

- 环戊醇衍生物的合成[2]
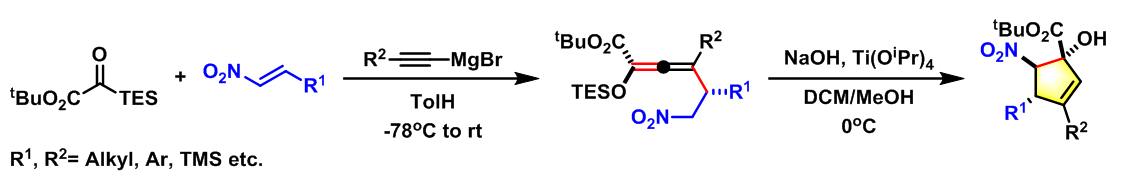
- 取代Indanone的对映选择性合成[3]
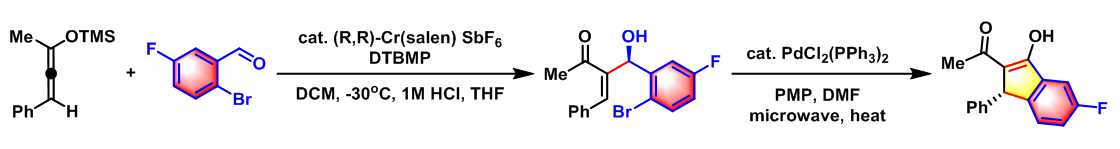


实验步骤
0°C下,向炔(1.1 eq.)的无水四氢呋喃溶液(底物浓度为0.64 M)中加入MeLi(1.1 eq., l:l LiBr配合物)的乙醚溶液(浓度为1.15 M)。将上述反应混合物搅拌15min后,将形成的溶液冷却至-78°C,再向该溶液中滴加三甲硅基酮(1 eq.),此时会形成白色沉淀。15 min后,再向上述反应混合物中加入亲电底物(1.3 eq.),之后,将上述反应混合物升温至0°C,继续搅拌至反应结束。反应结束后,采用冷的7% NaHCO3溶液淬灭反应,淬灭结束后,将反应液采用乙醚-正戊烷混合溶剂(1:l v/v)进行萃取,再依次用饱和食盐水进行洗涤、无水Na2SO4进行干燥。减压除去溶剂后,将残余物通过Kugelrohr蒸馏装置(24 mm,浴温89-91°C)进行蒸馏,获得相应目标产物。
实验技巧
参考文献
- [1]H. J.Reich, R. E. Olson, M. C.Clark, J. Am. Chem. Soc. 1980, 102, 1423.doi:10.1021/ja00524a036.
- [2]G. R. Boyce, S. Liu, J. S. Johnson, Org.Lett. 2012, 14, 652.doi:10.1021/ol2033527.
- [3]J. A.Brekan, T. E. Reynolds, K. A. Scheidt, J. Am.Chem. Soc. 2010, 132, 1472.doi:10.1021/ja909669e.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
通过在酰氧基酯与碱反应后产生2-羟基酮酯并伴随重排的反应。(参照标题图)
基本文献
- S. D. Lee, T. H. Chan, K. S. Kwon, Tetrahedron Lett., 1984, 25, 3399-3402.
DOI: 10.1016/S0040-4039(01)91030-5
反应机理

反应实例
R. A. Holton在进行紫杉醇的全合成时候,通过Chan重排反应将环状碳酸酯部分效率转化为γ内酯部分[1]。

除Holton这个例子以外,虽然再没有该反应用于全合成的实例。但是Reissig等人在Heliquinomycin的模型化合物的合成研究中,发现Chan重排作为副反应发生[2]。

实验步骤
实验技巧
参考文献
- R. A. Holton, C. Somoza, H. B. Kim, F. Liang, R. J. Biediger, P. D. Boatman, M. Shindo, C. C. Smith, S. Kim, H. Nadizadeh, Y. Suzuki, C. Tao, P. Vu, S. Tang, P. Zhang, K. K. Murthi, L. N. Gentile, J. H. Liu, J. Am. Chem. Soc., 1994, 116, 1597-1598.
DOI: 10.1021/ja00083a066 - C. Venkatesh, H.-U. Reissig, Synthesis, 2008, 22, 3605-3614.
DOI: 10.1055/s-0028-1083194
关联反应
概要
2级或者3级烷基醇通常在强酸条件下,发生重排反应形成乙烯酮(Meyer-Schuster重排)。特别是在末端具有乙炔基的叔醇,在类似反应条件下、给出具有不同取代方式的α,β-不饱和甲基酮的反应称为Rupe重排。该反应在碱性条件下也能发生。
基本文献
- Meyer, K. H.; Schuster, K. Ber. 1922, 55B, 819.
- Rupe, H.; Glenz, K. Ann. 1924, 436, 184.
- Rupe, H.; Kambli, E. Helv. Chim. Acta 1926, 9, 672. DOI: 10.1002/hlca.19260090185
- Swaminathan, S.; Narayanan, K. V. Chem. Rev. 1971, 71, 429. DOI: 10.1021/cr60273a001
反应机理
参考:J. Org. Chem. 1977, 42, 3403; J. Am. Chem. Soc. 1988, 110, 666.

反应实例(维基百科)
Meyer-Schuster反应有着广泛的应用,如以γ-硫取代烯丙醇为原料合成α,β-不饱和硫醚的过程中,需要使用PTSA作为催化剂将ω-炔基-ω-羟甲基内酯转化为烯酰胺[1];其他的应用还有如在温和酸性条件下3-炔基-3-羟基-1H-异吲哚重排为α,β-不饱和羰基化合物[2]。最有趣的一个应用是在合成紫杉醇的一个片段的过程中采用非对映异构的合成方法使得产物得到单一的E-烯烃[3]。

该步产率大于70%(如果计入在其他步骤中可以通过Meyer-Schuster转化的副产物则产率可达91%)。作者选用Meyer-Schuster重排的原因是他们希望在不破坏分子其余部分的前提下将一个有位阻的酮转化为烯。
实验步骤
实验技巧
参考文献
- Chihab-Eddine, A.; Daich, A.; Jilale, A.; Decroix, B. J. Heterocycl. Chem. 2000, 37, 1543-1548.(doi:10.1002/jhet.5570370622)
- Omar, E.A.; Tu, C.; Wigal, C.T.; Braun, L.L. J. Heterocycl. Chem. 1992, 29, 947-951.(doi:10.1002/jhet.5570290445)
- Crich, D.; Natarajan, S.; Crich, J.Z. Tetrahedron 1997, 53, 7139-7158.(doi:10.1016/S0040-4020(97)00411-0)
外部链接
- Meyer-Schuster Rearrangement (Wikipedia)
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
投稿作者:LiWen Xia
概要
1957年[1]. D.H.R.Barton 研究山道年的光照重排。
早在1902年,Francesconi等人将山道年溶解于醋酸水溶液中,在太阳光下照射几周,生成了一些产物,其中主要的产物,他们分离出来并命名为“isophotosantonin acid”。D.H.R.Barton在其基础上进行再次实验,将日光改为紫外灯照射。分离出“isophotosantonin acid”并且首次记录收率为30%以上。而后将“isophotosantonin acid”进行表征发现它并不是“isophotosantonin acid”而是 “isophotosantonin lactone”。
反应机理
结构确认后,Barton对其机理进行详细研究。
Barton研究的机理大致如下:
反应实例应用:
1.苦艾素(absinthin)全合成
2005年,上海有机所翟洪斌课题组将此反应应用于苦艾素(absinthin)的全合成中[2]。
从苦艾素的结构可以看出,其是二聚体,可以通过两分子的Diels-Alder反应得到。其单体结构 A就可以通过山道年光照重排来构建。

翟洪斌课题组路线如下:

- 2,5-环己二烯酮光照重排
2001年,Storn R. Jackson 等人[3]将此重排应用于环己二烯酮结构中。通过优化条件,S.R.Jackson发现在33%TFA in pentane 中能够得到82%收率的重排产物。

- (-)-Thapsigargin 全合成
2017年,应LEO Pharma 的要求,P.S.Baran [4]小组需要大量制备具有良好抗癌活性的(-)-Thapsigargin。前人工作 E.Stock等人需要用二氢香芹酮通过36-42步骤来合成(-)-Thapsigargin。Baran在(-)-Thapsigargin合成中应用光照环己二烯酮重排高效的构筑完(-)-Thapsigargin 的全部骨架。

最终以11步0.637%收率高效率得到(-)-Thapsigargin。同样的策略应用在十步合成(-)-Nortilobolide。
总而言之,从拓扑学意义来讲,山道年光照重排反应能够为构建具有五元环并七元环骨架的天然产物分子提供高效,简单的策略思路。

反应步骤
山道年光照重排实验操作[5]
氮气下,将5.00g(21mmol) α-santonin 溶解于400ml AcOH中,将反应瓶置于水冷却的石英砂中,温度控制在16℃。将反应体系置于150W的高汞灯下照射7小时。反应结束后将AcOH减压蒸除掉。将得到的油状液体溶解于20ml热甲醇中,后置于-20℃冷冻过夜。过滤得2.4g无色固体,收率38.6%。
参考文献
[1].D.H.R.Barton ,P.De.Mayo, Mohammed Shafiq., J .Chem. Soc. 1957. 929-935 DOI: 10.1039/JR9570000929 [2].Weihe Zhang, Shengjun Luo, Fang, Qingshou Chen, Hanwei Hu, Xueshun Jia, and Hongbin Zhai., J. Am. Chem. Soc. 2005, 127, 18-19. DOI: 10.1021/ja0439219 [3].Stona R. Jackson. Michael G. Johnson. Masafumi Mikami, Sojiro Shiokawa, and Erick M. Carreira., Angew. Chem. Int .Ed. 2001, 40, 2694. DOI: 10.1002/1521-3773(20010716)40:14<2694::AID-ANIE2694>3.0.CO;2-D [4].Hang Chu,Joel M. Smith, Jakob Felding, and Phil S.Baran., ACS Cent. Sci. 2017, 3, 47-51. DOI: 10.1021/acscentsci.6b00313 [5]. Weihe Zhang, Shengjun Luo, Fang, Qingshou Chen, Hanwei Hu, Xueshun Jia, and Hongbin Zhai., J. Am. Chem. Soc. 2005, 127, 18-19. DOI: 10.1021/ja0439219
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
1,5-dien-3-ol型化合物、发生与Cope Rearrangement同样的[3,3]sigmatropic重排,形成δ,ε-不饱和糖基化合物。Cope重排是可逆的平衡反应,生成物的比值根据其热力学的稳定性决定。而Oxy-Cope重排反应却有不同、中间体烯醇会发生异构化形成更稳定的酮化合物,因此几乎是不可逆的形成产物。
Evans等人发现,通过碱性条件,拔去OH的质子形成氧负离子,可以使得重排反应发生剧烈加速(大概速率提高1010-1017)。这被称为Anionic Oxy-Cope重排。具有高分极度的醇盐具有较大的反应加速效果,并且使用KH和18-冠醚-6的组合可以加速反应的进行。由于该反应不需要高温,因此同样适用于热不稳定的化合物。
关联反应(本网站)
- Ichikawa Allylcyanate Rearrangement
- Vinylcyclopropane Rearrangement
- Overman Rearrangement
- Aza-Cope Rearrangement
- Eschenmoser-Claisen Rearrangement
- Cope Rearrangement
- Johnson-Claisen Rearrangement
- Ireland-Claisen Rearrangement
- Wittig Rearrangement
- Claisen Rearrangement
基本文献
- Berson, J. A.; Jones, M., Jr. J. Am. Chem. Soc. 1964, 86, 5017. DOI: 10.1021/ja01076a066
- Berson, J. A.; Jones, M., Jr. J. Am. Chem. Soc. 1964, 86, 5019. DOI: 10.1021/ja01076a067
- Anionic oxy-Cope: Evans, D. A.; Golob, A. M. J. Am. Chem. Soc. 1975, 97, 4765. DOI: 10.1021/ja00849a054
- Rhoads, S.J.; Raulins, N. R. Org. React. 1975, 22, 1.
反应机理
与其他的[3,3]-sigmatropic重排反应一样、经由6元环过渡态(普通情况下是椅式)进行反应。具有潜不对称点的底物,不对称可以反应到产物中。

反应实例
Periplanone B 的合成[1]:形成10元环手性化合物。

(+)-CP-263,114的合成[2] :Oxy-Cope重排组合使用进行串联反应,高效率合成anti-Bredt型双环化合物。

实验步骤
实验技巧
参考文献
[1] (a) Still, W. C. J. Am. Chem. Soc. 1979, 101, 2493. DOI: 10.1021/ja00503a048(b) Adams, M. A.; Nakanishi, K.; Still, W. C.; Arnold, E. V.; Clardy, J.; Persoons, C. J. J. Am. Chem. Soc. 1979, 101, 2495. DOI: 10.1021/ja00503a049[2] Chen, C.; Layton, M. E.; Sheehan, S. M.; Shair, M. D. J. Am. Chem. Soc. 2000, 122, 7424. DOI: 10.1021/ja001958x
外部链接
- Cope Rearrangement (oxy-Cope)
- Cope Rearrangement (Wikipedia)
- Cope Rearrangement (organic-chemistry.org)
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!