本文作者:杉杉
导读
近日,美国Colorado州立大学 (Colorado State University)的J. S. Bandar课题组报道一种通过Lewis碱性盐试剂促进的苄基三甲基硅烷与(杂)芳基腈、砜以及氯代之间的交叉偶联反应方法学,进而获得一系列1,1-二芳基烷烃衍生物。值得注意的是,这一全新的偶联策略中,选择稳定、廉价并易于制备的有机硅烷作为偶联参与物,同时,能够在酸性官能团的存在下,实现选择性的芳基化反应。此外,该小组通过对这一新型的偶联策略在药物类似物分子的合成以及多组分反应中的应用,进一步阐明这一全新的偶联反应方法学具有良好的合成应用价值。
Lewis Basic Salt-Promoted Organosilane Coupling Reactions with Aromatic Electrophiles
T. W. Reidl, J. S. Bandar, J. Am. Chem. Soc. 2021, 143, 11939. doi: 10.1021/jacs.1c05764.

正文
1,1-二芳基烷烃是一类具有较高应用价值的有机分子,通常采用官能团化的苄基试剂与芳香亲电底物之间的偶联反应策略,完成上述分子的制备。其中,过渡金属催化的芳卤或芳基拟卤代物与苄基镁、锌以及硼化合物之间的偶联反应方法学应用最为广泛。然而,上述策略中,通常需要原位制备出具有较高反应活性的苄基试剂,因此,实验操作较为繁琐。由此,有机合成化学家开始致力于设计全新的偶联反应策略,进而实现1,1-二芳基烷烃分子的高效构建。
苄基去质子化反应方法学[1]作为一种极具吸引力的反应策略,能够通过碳负离子中间体的形成,进而与芳基亲电底物在金属催化剂存在或无催化剂存在的条件下,完成相应的偶联反应过程 (Figure 1, left)。然而,这一策略中,却伴随多芳基化副产物的产生,并且,需要选择具有一定酸性的前亲核底物,例如二芳基甲烷。同时,上述去质子活化过程的底物应用范围同样较为有限。
为解决上述问题,本课题组开始致力于设计一种采用碱促进的全新的苄位芳基化策略,反应过程中涉及Lewis碱对于具有一定Lewis酸性的苄基化合物的活化,进而实现芳基Csp2-Csp3键之间的偶联过程[2]。其中,由于苄基三甲基硅烷具有良好的空气稳定性、无吸湿性以及廉价易得等优势,由此成为较为理想的偶联底物。以此同时,涉及复杂苄基三甲基硅烷合成[3]的相关反应策略已有文献报道。而且,迄今为止,由于苄基三甲基硅烷分子所具有的高度稳定性,致使其在金属催化的交叉偶联反应过程中,无法表现出良好的反应活性,进而使其在芳基化反应方法学中的应用受到较大限制[4]。为进一步解决上述反应过程中存在的局限,同样有文献报道采用钯催化以及金属光氧化还原催化[5]的相关偶联反应策略 (Figure 1, right)。
这里,J. S. Bandar课题组报道一种全新的,通过Lewis碱性盐试剂促进的苄基三甲基硅烷与各类芳香亲电底物之间的直接偶联反应方法学,进而获得一系列相应的1,1-二芳基烷烃衍生物 (Figure 1, bottom)。值得注意的是,在这一全新的偶联策略中,苄位的芳基化能够与潜在的负离子副反应进行有效的竞争,进而使这一新型的偶联反应过程,能够与底物中存在的酸性与亲电官能团良好地进行兼容。并且,同样能够良好地应用于其它硅烷底物以及其它相关的反应序列,例如采用烯丙基硅烷参与的串联芳基化/异构化反应方法学,能够较好地完成各类芳基烯分子的构建。因此,采用Lewis碱促进的芳基化反应方法学,具有良好的合成实用性以及较为广泛的底物适用范围。

同时,在2019年,J. S. Bandar团队报道一种通过氟离子活化的烯丙基三甲基硅烷与三氟甲基芳烃之间的单选择性去氟烯丙基化反应方法学 (monoselective defluoroallylation)[6]。其中,反应过程涉及负离子烯丙基中间体的形成,并进一步与三氟甲基芳烃经历单电子转移过程,进而使C-F键断裂,并通过形成的二氟苄基自由基,进行后续的烯丙基化过程 (Scheme 1a)。这一策略类似于采用烯丙基三甲基硅烷进行的1,4-二氰基芳烃的光诱导电子转移 (PET)烯丙基化过程,即反应机理中,SET过程先于C-C键的形成过程 [7]。同时,在PET过程的研究中,该小组同样对苄基三甲基硅烷的应用进行研究。然而,上述的PET烯丙基化过程具有较低程度的区域选择性以及副产物形成的问题,同时,反应过程中需要进一步采用紫外光辐射 (Scheme 1b) [8]。基于上述的研究报道,作者假设,有机三甲基硅烷通过Lewis碱的活化,能够良好地促进其与其它芳香亲电底物之间的直接偶联过程。
为进一步验证这一假设,作者选择4-氰基吡啶 (1)与苄基三甲基硅烷 (2) 作为模型底物,对相应的Lewis碱进行筛选。最终,该小组确定出最佳的反应条件为:采用1 eq.的氟化铯为碱,同时加入1.1 eq.18-crown-6,在DMSO作为反应溶剂以及室温的反应条件下,反应时间为3 h,进而获得95%收率的偶联产物3 (Scheme 1c)。

在上述的最佳反应条件下,作者首先对上述偶联过程的底物应用范围进行考察 (Table 1)。研究表明,缺电子的氰基吡啶与氰基苯底物能够与一系列一级、二级以及三级苄基硅烷在上述的标准反应条件下,进行相应的偶联过程,并获得最终的目标产物4–24,收率为50-94%。同时,反应过程中表现出优良的官能团兼容性。之后,该小组观察到,具有α-杂原子的苄基硅烷底物 (25–27)以及药物分子paroxetine (28)与bepotastine (29),均能够与上述的标准反应条件良好地进行兼容。

之后,作者对各类芳基亲电底物的应用范围进行考察 (Scheme 2)。研究表明,2-氯-1,3-唑类为上述偶联过程的有效底物 (30–32)。并且,具有扩张π-体系的氯代物,例如1,3-二氯异喹啉 (33)、9-氯吖啶(34)以及抗肿瘤药物分子imiquimod的氯喹啉衍生物 (35)同样能够与上述的最佳反应条件良好地兼容。同时,作者发现,尽管4-卤吡啶在上述的标准反应条件下,无法有效地进行相应的偶联过程,然而,上述的标准反应体系对于4-磺酰基吡啶 (36)底物,则能够获得具有良好收率的偶联产物 (Scheme 2b)。此外,作者观察到,将4-氯吡啶37转化为磺酰化产物38 (Scheme 2c)之后,再与苄基硅烷进行上述的偶联过程,最终能够顺利获得相应的二芳基烷烃产物39。


接下来,作者进一步对有机硅化合物40与各类芳基与杂芳基亲电底物之间的偶联过程进行进一步研究 (Figure 2)。该小组观察到,通过化合物40参与的偶联过程,最终能够获得一系列相应的chlorpheniramine类似物,例如41–48,收率为45-95%。

此外,作者还同样对上述偶联过程的选择性进行深入研究 (Scheme 3)。首先,作者发现,芳基亲电底物2在上述的标准反应条件下,能够获得80%收率的甲苯产物 (Scheme 3a)。进而表明苄基的质子化过程与芳基化过程之间存在竞争。然而,较为有趣的是,在酸性显著高于甲苯的反应溶剂中,4-氰基吡啶的苄基化过程同样能够较好地进行 (Scheme 3b)。此外,作者在选择苄基硅烷的两种区域异构体 (50与52) 参与的偶联过程中发现,上述苄基硅烷的芳基化过程能够区域专一性地在具有TMS基团的位置进行 (Scheme 3c)。进而表明,芳基化过程在质子转移之前进行。同时,研究发现,具有一定酸性的二芳基烷烃产物的去质子化过程受到极大程度的抑制,进而能够进一步抑制多芳基化副反应过程的进行。上述事实同样表明,Lewis碱促进的芳基化反应方法学具有显著的优势,而在Brønsted碱参与的芳基化过程中,则无法形成相应的苄基碳负离子。同时,可能出现多重芳基化的副反应。并且,在具有多重苄基位置取代的相关底物中,无法获得较为优良的选择性 (详见Supporting Information)。

为解释上述芳基化过程中的高度选择性,作者假设苄基负离子中间体通过极性或SET机理,进行快速的芳香取代过程 (Figure 3)。其中,SET机理过程通过碱性条件促进,并类似于有机硅烷与1,4-二氰基芳烃之间PET反应。并且,极性机理同样具有合理性,因为氰基芳烃与磺酰基芳烃分子能够参与通过典型的加成-消除过程进行的取代反应。而在负离子试剂对于类似亲电底物的加成过程中,准确区分上述两种不同的机理路径,则具有较大的挑战性。并且,上述的偶联反应过程中的相关实验结论,均能够通过极性与SET两种不同的机理路径进行合理的解释。同时,作者发现上述的偶联反应方式可能具有相关的底物依赖性,尽管相应的芳基化过程能够与其它通过潜在负离子中间体引发的副反应过程有效地进行竞争。

通过上述研究,作者设想,有机硅烷的芳基化过程同样能够与碱促进的其它合成转化过程进行有效地结合 (Scheme 4)。首先,作者发现,通过烯丙基三甲基硅烷形成的烯丙基芳烃中间体,能够进一步经历后续的立体选择性异构化过程,并获得相应的烯产物54、55与56 (Scheme 4a)。之后,该小组进一步设计出一种有机硅烷、芳基亲电底物与Michael受体之间的三组分偶联策略。作者假设反应过程中,首先进行有机硅烷与芳基亲电底物之间选择性的苄基芳基化步骤,之后,通过剩余有机硅烷与负离子的结合,进而引发后续的Michael加成反应 (Scheme 4b)。同时,作者发现,γ,γ-二芳基酰胺57与58同样能够通过上述的三组分反应策略进行有效的制备。之后,该小组观察到,采用甲基烯丙基三甲基硅烷底物时,能够通过碱促进的三步选择性的反应过程 (即芳基化、加成与烯基异构化),最终形成四取代烯基化合物59。

总结
美国Colorado州立大学的J. S. Bandar课题组报道一种通过Lewis碱性盐试剂促进的苄基以及烯丙基三甲基硅烷与(杂)芳基腈、砜以及氯代物之间的偶联反应方法学。并且,通过这一全新的偶联策略,能够以优良的区域选择性与单选择性 (monoselectivity),获得一系列1,1-二芳基烷烃以及芳基烯衍生物。同时,上述策略与现有的合成转化方法学形成有效的互补。此外,该小组通过上述偶联策略在药物类似物分子的合成以及在多组分反应中的应用,进一步阐明这一全新的偶联反应方法学具有良好的合成应用前景。
参考文献
[1] (a) M. Li, S. Berritt, L. Matuszewski, G. Deng, A. Pascual-Escudero, G. B. Panetti, M. Poznik, X. Yang, J. J. Chruma, P. J. Walsh, J. Am. Chem. Soc. 2017, 139, 16327. doi: 10.1021/jacs.7b09394.(b) X. Ji, T. Huang, W. Wu, F. Liang, S. Cao, Org. Lett. 2015, 17, 5096. doi: 10.1021/jacs.7b09394.
[2] M. Takeda, K. Nagao, H. Ohmiya, Angew. Chem. Int. Ed. 2020, 59, 22460. doi: 10.1002/anie.202010251. [3] (a) S. Utsumi, T. Katagiri, K. Uneyama, Tetrahedron 2012, 68, 1085. doi: 10.1016/j.tet.2011.11.082.(b) P. K. Kundu, S. K. Ghosh, Tetrahedron 2010, 66, 8562. doi: 10.1016/j.tet.2010.09.001.
[4] (a) Y. Wu, S. Bouvet, S. Izquierdo, A. Shafir, Angew. Chem. Int. Ed. 2019, 58, 2617. doi: 10.1002/anie.201809657.(b) J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu, Q. Wang, Org. Chem. Front. 2019, 6, 2902. doi: 10.1039/C9QO00690G.
(c) M. Puthanveedu, V. Polychronidou, A. P. Antonchick, Org. Lett. 2019, 21, 3407. doi: 10.1021/acs.orglett.9b01141.
[5] (a) K. Itami, M. Mineno, T. Kamei, J. Yoshida, Org. Lett. 2002, 4, 3635. doi: 10.1021/ol026573t.(b) V. Corcé, L.-M. Chamoreau, E. Derat, J.- P. Goddard, C. Ollivier, L. Fensterbank, Angew. Chem. Int. Ed. 2015, 54, 11414. doi: 10.1002/anie.201504963.
[6] C. Luo, J. S. Bandar, J. Am. Chem. Soc. 2019, 141, 14120. doi: 10.1021/jacs.9b07766. [7] Y. Liu, H. Li, S. Chiba, Org. Lett. 2021, 23, 427. doi: 10.1021/acs.orglett.0c03935. [8] Y. Deng, Q. Liu, A. B. Smith, III. J. Am. Chem. Soc. 2017, 139, 9487. doi: 10.1021/jacs.7b05165.本文作者:杉杉
导读
含氮-氮键的化合物,广泛存在于生物活性化合物、有机材料等中。然而,对于分子间的选择性N-N氧化偶联反应,仍具有难度。近日,德国亚琛工业大学Frederic W. Patureau课题组在Organic Letters 上发表论文,报道了一种通过使用高价碘作为末端氧化剂,实现酰胺和苯并三唑的选择性交叉脱氢N-N偶联反应。
Cross-Dehydrogenative N-N Coupling of Aromatic and Aliphatic Methoxyamides with Benzotriazoles
Pooja Y. Vemuri and Frederic W. Patureau*
Org. Lett. 2021, 23, 3902–3907. DOI:10.1021/acs.orglett.1c01034

正文
含有氮-氮键的化合物,广泛存在于药物、天然产物、有机材料、染料等中(Figure 1)。然而,此类化合物的合成常依赖重氮化反应或使用亲电的氮源(如肟、腙、腈、叠氮化物等),但对于通过直接脱氢构建N-N键的策略却很少被研究。

由于步骤和原子经济性的特点,脱氢交叉偶联(CDC)反应备受关注,同时可避免底物的预活化,并以高选择性构建C-C、C-N、C-O、C-P、C-S和杂SA-SB键(Scheme 1)。在过去,脱氢N-N偶联反应常用于分子内N-N键的构建。然而,对于分子间的脱氢N-N偶联反应,仍未得到充分探索。据文献查阅,Stahl等[1]和Jin等[2]报道了分子间脱氢N-N偶联的方法,但仅限于咔唑与二芳基胺底物(Scheme 1)。在此,本文将报道一种通过简单且温和的反应条件,实现酰胺和苯并三唑的高度选择性脱氢偶联反应。

首先,作者以N-甲氧基苯甲酰胺1a和苯并三唑2a作为模型底物,进行了相关偶联反应条件的筛选(Table 1)。反应的最佳条件为:以PIDA为氧化剂,六氟异丙酚(HFIP)为溶剂,可在40 °C下反应,获得73%收率的偶联产物3aa。

在获得上述最佳反应条件后,作者开始对底物范围进行了扩展(Scheme 2)。酰胺底物中的芳基取代,不受电子效应和定位效应的影响,均可顺利反应,获得相应的产物3aa–3ra,收率为40-73%。同时,对于对称的苯并三唑,也获得产物3ab、3ob和3ac。对于非对称的苯并三唑,通常获得异构体的混合物,如3ad/3ad’–3aj/3aj’。此外,一系列脂肪族酰胺(3sa,3sb,3ta,3ua),也与体系兼容。


随后,作者进行了一些竞争性实验,以探究每种偶联底物的相对亲核性(Scheme 3)。酰胺1b与苯并三唑2b和2c同时反应时,具有给电子取代基的苯并三唑收率略高(3bb/3bc = 1.3)。苯并三唑2a与酰胺1b和1f同时反应时,具有给电子取代基的酰胺收率略高(3ba/3fa = 1.9)。上述结果说明,具有给电子基的底物反应速度会偏快,收率也略高。

同时,为了进一步证明反应的先后顺序,作者分别将底物分批加入至反应体系中(Scheme 4)。在Experiment A中,酰胺底物1a在标准条件反应16 h后,再加入苯并三唑2a,但未能获得目标产物3aa。在Experiment B中,苯并三唑2a在标准条件反应16 h后,再加入酰胺底物1b,可获得61%收率的目标产物3ba。这些结果表明,在PIDA存在下,酰胺底物会发生不可逆地降解,而苯并三唑则不受影响。此外,作者还提出了一种可能的反应机理(Scheme 5)。

为了进一步证明反应的实用性,作者进行了相关的克级反应(Scheme 6),克级实验结果表明,仅需将反应温度提高至60 °C,即可获得60%收率的目标产物3aa。

此外,合成的产物可应用于C-H活化偶联反应中(Scheme 7)。当使用Rh催化剂时,3aa可与二苯基乙炔顺利进行偶联反应,获得60%收率的化合物4。值得注意的是,化合物4未含有氮原子,从而表明这种新型O-N-N-N-N官能团可作为金属催化C-H键活化反应中有效的离去基团。

总结
德国亚琛工业大学Frederic W. Patureau课题组报道了一种N-甲氧基酰胺和苯并三唑的分子间脱氢N-N偶联反应。同时,该反应具有反应条件温和、底物范围广泛、官能团耐受性高等特点。值得注意的是,产物中的O-N-N-N-N官能团,可作为金属催化C-H键活化反应中有效的离去基团。
参考文献
[1] (a) Ryan, M. C.; Martinelli, J. R.; Stahl, S. S. Cu-Catalyzed Aerobic Oxidative N−N Coupling of Carbazoles and Diarylamines Including Selective Cross-Coupling. J. Am. Chem. Soc. 2018, 140, 9074. (b) Ryan, M. C.; Kim, Y. J.; Gerken, J. B.; Wang, F.; Aristov, M. M.; Martinelli, J. R.; Stahl, S. S. Mechanistic insights into coppercatalyzed aerobic oxidative coupling of N−N bonds. Chem. Sci. 2020, 11, 1170. See also: (c) Monir, K.; Ghosh, M.; Mishra, S.; Majee, A.; Hajra, A. Phenyliodine(III) Diacetate (PIDA) Mediated Synthesis of Aromatic Azo Compounds through Oxidative Dehydrogenative Coupling of Anilines: Scope and Mechanism. Eur. J. Org. Chem. 2014, 2014, 1096. [2] For a hypervalent iodine oxidizing system, see: Yin, D.; Jin, J. Transition-Metal-Free Dehydrogenative N−N Coupling of Secondary Amines with KI/KIO4. Eur. J. Org. Chem. 2019, 2019, 5646.本文作者:杉杉
导读
近日,加州大学伯克利分校John F. Hartwig教授课题组在ACS Catalysis发表论文,报道了一种简单芳烃与芳基溴化物直接芳基化的策略。在该体系中,无需使用任何导向基团,借助银-钯双金属的协同催化循环(银配合物裂解芳烃C-H键,而钯催化剂能够形成联芳基产物),即可实现相关的偶联过程。此外,通过动力学同位素效应、竞争实验和氢-氘交换实验进一步对反应机理进行了研究,其中银配合物催化C-H键的裂解为决速步骤。
Direct Arylation of Simple Arenes with Aryl Bromides by Synergistic Silver and Palladium Catalysis
Adrian Tlahuext-Aca, Sarah Yunmi Lee, Shu Sakamoto, and John F. Hartwig*
ACS Catal. ASAP, DOI:10.1021/acscatal.0c05254

正文
碳氢键的功能化作为从易获得原料构建碳-碳和碳-杂原子键的有效方案。其中,芳烃与芳基亲电试剂的直接芳基化可直接实现碳-芳基键的构建,从而合成医药、农药和有机材料中常见的联芳基骨架(Scheme 1a)。尽管已报道多种直接芳基化的催化方法,但无导向基团的简单芳烃的分子间芳基化仍具有挑战。目前,对于在无过渡金属催化剂和导向基团时,直接芳基化的方法需过量的芳烃,或者含有几个可促进C-H活化的给电子或吸电子基团的底物(Scheme 1b, left)。最近,Larrosa等[1]报道了一种简单芳烃直接芳基化的策略,其中Cr(CO)3与芳烃配位促进了芳基C-H键的活化,但是该方法需要预先引入Cr(CO)3,并在后期进行脱除(Scheme 1b, right)。基于前期钯和银催化简单芳烃的烯丙基化的相关成果[2],在此,加州大学伯克利分校John F. Hartwig教授课题组报道了一种过渡金属催化芳基溴化物与普通芳烃(1.5-5.0 eq)的直接芳基化反应。同时,通过一系列动力学数据(如动力学同位素效应、竞争实验和H/D交换反应)表明,反应涉及银配合物催化促进芳烃C-H键的活化以及钯催化[3]促进芳基溴化物氧化加成的过程(Scheme 1c)。

首先,作者以1-氟萘1a和3-溴甲苯2a作为模型底物,进行了相关偶联反应条件的筛选(Table 1)。反应结果表明,在反应温度为120℃的条件下,当以Ag2O为银盐、Pd(OAc)2为催化剂、Cy2PtBu为配体,Cs2CO3为碱时,可在tAmOH溶剂中反应,获得81%收率的目标产物3aa。

在获得上述最佳反应条件后,作者开始对芳烃的范围进行了扩展(Scheme 2)。当以4-氯(或三氟甲氧基)苯甲醚为底物时,可获得高收率等摩尔异构体的混合物3ba–3ca,但以4-硝基苯甲醚为底物时,获得收率较低的产物3da(具有高选择性)。同时,当以4-氯三氟甲苯和2,2-二氟苯并二噁唑为底物时,反应平稳进行,以高收率和优异的选择性形成联芳基产物3eb和3fa。此外,各种氟代芳烃底物均与体系兼容,从而获得相应的产物3ga–31a和3mb。

紧接着,作者研究了1-氟萘(1a)与不同取代芳基溴化物的范围(Scheme 3)。在对位和间位含有供电子基团的芳基溴化物,如甲氧基、吗啉基、苯氧基和硫代甲氧基,可获得中等收率的联芳基产物3ab–3ae。而在间位和对位含有吸电子基团的芳基卤化物,仅获得较低收率的联芳基产物3af–3ah。此外,3,4-二取代的芳基溴化物也可顺利反应,以高收率合成了3ai。同时,空间位阻更大的底物,也可顺利进行反应,获得产物3aj–3al。

为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 4)。首先,通过2a与1g和[2H] 1g的直接芳基化的初始速率对比发现,KIE值为4.0±0.3,从而说明芳烃C-H键的裂解为决速步骤(Scheme 4a)。同时,通过1k和1h之间的竞争实验表明,缺电子芳烃1k的反应性更高,这与通过金属化去质子化步骤进行C-H键断裂的机理一致[4]。

此外,作者以1,3-二氟苯(1l)和10当量的D2O在氘代叔戊醇中进行了H/D交换的对比性实验(Table 2)。当在0.5当量的Ag2O,40% mol的tBuPCy2和化学计量的Cs2CO3存在下,获得84%的氘掺入2-位的[2H] 11(entry 1)。而在Pd(OAc)2存在下,氘在1l中的掺入率较低(entry 2)。在没有Ag2O或配体时,仅进行少量的H/D交换(entries 3-4)。此外,在没有银添加剂时,Pd(OAc)2不能促进1l的H/D交换。这些结果表明,在芳基化反应的速率确定步骤中,银配合物会裂解芳烃的C-H键。

总结
加州大学伯克利分校John F. Hartwig教授课题组报道了一种简单芳烃与市售芳基溴化物直接芳基化的策略,涉及银和钯协同催化体系。同时,该方法无需任何导向基团,具有广泛的底物范围,并且使用较低的芳烃的量。此外,机理研究表明,银配合物裂解芳烃的C-H键,可能是通过协同的金属化-去质子化步骤进行,同时作为决速步骤。
参考文献
[1] (a) Ricci, P.; Krämer, K.; Cambeiro, X. C.; Larrosa, I. Arene-Metal π-Complexation as a Traceless Reactivity Enhancer for C-H Arylation. J. Am. Chem. Soc. 2013, 135, 13258-13261. (b) Ricci, P.; Krämer, K.; Larrosa, I. Tuning Reactivity and Site Selectivity of Simple Arenes in C-H Activation: Ortho-Arylation of Anisoles via Arene-Metal π-Complexation. J. Am. Chem. Soc. 2014, 136, 18082-18086. (c) Whitaker, D.; Burés, J.; Larrosa, I. Ag(I)-Catalyzed C-H Activation: The Role of the Ag(I) Salt in Pd/AgMediated C-H Arylation of Electron-Deficient Arenes. J. Am. Chem. Soc. 2016, 138, 8384-8387. (d) Whitaker, D.; Batuecas, M.; Ricci, P.; Larrosa, I. A Direct Arylation-Cyclization Reaction for the Construction of Medium-Sized Rings .Chem. – Eur. J. 2017, 23, 12763-12766. (e) Batuecas, M.; Luo, J.; Gergelitsová, I.; Krämer, K.; Whitaker, D.;mVitorica-Yrezabal, I. J.; Larrosa, I. Catalytic Asymmetric C-H Arylation of (η6-Arene)Chromium Complexes: Facile Access to Planar-Chiral Phosphines. ACS Catal. 2019, 9, 5268-5278. (f) Panigrahi, A.; Whitaker, D.; Vitorica-Yrezabal, I. J.; Larrosa, I. Ag/PdCocatalyzed Direct Arylation of Fluoroarene Derivatives with Aryl Bromides.ACS Catal. 2020, 10, 2100-2107. [2] Lee, S. Y.; Hartwig, J. F. Palladium-catalyzed, Site-selective, Direct Allylation of Aryl C-H Bonds by Silver-mediated C-H Activation: A Synthetic and Mechanistic Investigation. J. Am. Chem. Soc. 2016, 138, 15278-15284. [3] For other dual Ag/Pd catalytic systems: (a) Lotz, M. D.; Camasso, N. M.; Canty, A. J.; Sanford, M. S. Role of Silver Salts in Palladium-Catalyzed Arene and Heteroarene C-H Functionalization Reactions. Organometallics 2017, 36, 165-171. (b) Colletto, C.; Panigrahi, A.; Fernandez-Casado, J.; Larrosa, I. Ag(I)-C-H Activation Enables Near-Room-Temperature Direct α-Arylation of Benzo[b]thiophenes. J. Am. Chem. Soc. 2018, 140, 9638-9643. (c) Li, W.; Yuan, D.; Wang, G.; Zhao, Y.; Xie, J.; Li, S.; Zhu, C. Co-operative Au/Ag Dual-Catalyzed Cross-Dehydrogenative Biaryl Coupling: Reaction Development and Mechanistic Insight. J. Am. Chem. Soc. 2019, 141, 3187-3197. (d) Lee, J. A.; Luscombe, C. K. Dual-Catalytic Ag-Pd System for Direct Arylation Polymerization to Synthesize Poly(3-hexylthiophene). ACS Macro Lett. 2018, 7, 767-771. [4] (a) Lapointe, D.; Fagnou, K. Overview of the Mechanistic Work on the Concerted Metallation-Deprotonation Pathway. Chem. Lett. 2010, 39, 1118-1126. (b) Carrow, B. P.; Sampson, J.; Wang, L. Base-Assisted C-H Bond Cleavage in Cross-Coupling: Recent Insights into Mechanism, Speciation, and Cooperativity. Isr. J. Chem. 2020, 60, 230-258.本文来自Chem-Station日文版 第一手はこれだ!:古典的反応から最新反応まで3 |第8回「有機合成実験テクニック」(リケラボコラボレーション) webmaster
翻译投稿 炸鸡 校对 HaoHu
这次和理学系实验室网站合作,我们会陆续推出十篇的“有机合成实验技巧”特集。
从第六篇开始将持续为期三篇的“合成化学的第一步!:找遍古典反应到最新反应”系列,这是“合成化学的第一步!:找遍古典反应到最新反应”系列的第三篇,将教大家如何选择实践中用到的合成反应的实验条件。第六篇和第七篇已经为大家介绍了氧化反应・保护基的脱除,还有缩合反应等有机合成化学中常用反应的首选。第八篇为大家介绍交叉偶联反应,内容不仅考虑到了有机合成专业的人员也考虑到了非有机合成专业的人员。
交叉偶联反应的基础
请读者看下面的反应式。这是笔者给大学3年级学生教授有机金属化学的时候举的一个非常有代表性的偶俩反应——铃木—宫浦交叉偶联反应。
当有机硼化合物和有机卤化物在磷酸钾,xPhos配体和Pd2dba3催化剂存在下反应时,可以90%的产率获得偶联产物。反应机理为:有机卤化物的C–Cl键被氧化加成到钯,与硼化合物发生金属转移,随后发生还原消除。

对这个反应完全不知道的同学会很难理解本篇内容,建议这类同学再复习下有机金属化学再来阅读本篇内容。对于那些不明白交叉偶联反应但是想做的同学可以借鉴本篇内容。有些同学明白反应但是却不懂为什么要用这么复杂的试剂和反应条件的同学,建议你们阅读整篇文章。在我教授有机金属化学的课程时,学生们普遍都有以下五个疑问:
- 1.为什么反应物组合得是有机硼化合物和有机卤化物呢?
- 2.为什么要用钯作为催化剂呢?
- 3.配体是怎么影响反应的?
- 4.碱是怎么在反应起作用的?
- 5.反应机理是什么?氧化加成/金属转移/还原消除的机理?
有上述疑问的同学不妨仔细看看有机化学课本上的有关交叉偶联反应历史的介绍,相信你们看完了交叉偶联反应的历史就会明白上述反应条件的含义了。
即使在化学领域,也有许多研究几乎不涉及有机合成。但是,我想让您记住获得诺贝尔化学奖的交叉偶联反应,如果您从事化学研究,那么这个反应的使用频率就会非常的高。
交叉偶联反应的首选反应条件是什么呢?
说了这么多,那么我们本次的主角——交叉偶联反应(铃木—宫浦反应)的首选反应条件是什么呢?
对于偶联反应的初学者来说,偶联反应的反应条件很多,不知道该选择哪一个。当然你可以用SciFinder检索你需要做的实验的反应条件和各种反应底物,如果能找到的反应物就是自己想做的或者和想做的反应物类似而且用到的试剂不是很复杂,价格不是特别昂贵的话就采用那个反应条件了。
但是如果检索不到类似的反应物,但是得立马得做偶联反应那该怎么办呢?
遇到这种情况的话请试试下面的条件。
乙酸钯,三苯膦,碳酸钠,甲苯/水(4:1),80度
从笔者的经验上看这是能保证反应顺利进行的且成本最低的一个反应条件。如果更改了碱,反应变化会很大,如果反应没有进行,试试把碱换成碳酸钾,磷酸钾等。

偶联反应最便宜的首选反应条件
当然不可否认比这个好的反应条件有很多。我下面介绍的是除了这个首选反应条件之外的一些反应条件,我们可以尝试更改一些条件来获得不一样的反应条件(没有学术讨论的意味)。
钯盐:氯化钯或溶解性稍微好一点的氯化钯乙腈络合物,再有Pd(dba)2之类的也可以试试。
配体:三苯膦是最便宜的,但是如果用三苯膦氧化加成无法进行时,请尝试使用体积大一点的烷基膦(Cy3P。TBu3P)。万能的Buchwald配体可用在很多地方。在众多的Buchwald配体中,推荐读者使用XPhos配体,因为它非常好用。

Xphos
添加剂:在某些情况下,添加冠醚或氯化锂可以显着提高产率。
溶剂:也可以尝试使用THF/H2O溶剂体系和二噁烷/水溶剂体系。
不涉及官能团的交叉偶联反应
以上的内容或许对从事有机合成研究的人来说是不够的。所以我再稍微介绍下近年流行的碳氢键直接活化偶联反应的首选。
上述交叉偶联反应中,偶联剂中需要有官能团存在,例如卤素或硼酸,并且必须进行几个额外的反应以引入官能团。如果可以在不用引入一方或两方偶联剂的官能团的情况下进行偶联反应,则实验效率就会翻倍。这种反应在近十年十分流行,有很多这样的反应条件相继被报道出来。
例如,1,3-唑化合物的芳基化反应。这个反应是笔者比较擅长的一个交换反应。在尝试了各种金属,配体和芳基化剂后,探索出的最佳的反应条件如下:
乙酸钯,三苯膦,碳酸铯,DMF,140度[1]

这个反应条件是由大阪大学的三浦教授团队报道出来的初期反应条件,尽管之后有很多新反应条件出现但这个反应条件还是很好用。配体是三苯基膦,非常便宜。当然最好在反应进行后考虑最新的反应条件以进行进一步优化,但三浦教授开发的反应条件足以当做首选了。切记从一开始就千万不要使用廉价的镍或其他的芳基化剂。
接下来是C3位(β位)芳基化反应生成苯并噻吩和噻吩。作者先前曾报道过类似类型的反应[2a],但这是首选反应条件。
芳基碘・Pd2(dba)3・Ag2CO3, HFIP

这个反应条件是由英国曼彻斯特大学的Larrosa报道的[3]。美中不足的是HFIP有点小贵,但是该反应条件只要室温就可以了。虽然笔者自己报道的反应条件[2b]也不差,但为了做出更好的实验成果还是用了其他人开发的反应条件。
最后,是苯基吡啶的芳基化反应。Ackermann报道的下图的Rh催化反应是首选[4]。

当偶联反应进行的不顺利时不妨试试下面这个反应条件,这个反应条件含有钴催化剂。虽然要加入格氏试剂,但这个反应不需要加热,在室温下就能进行。

自己试试看吧!
以上就是有关交叉偶联反应的反应条件的首选的介绍。至此,我们为期3回的“合成化学的第一步!”到这里也就结束了。如果你手上有最佳的首选反应条件,那么就能很快找到需要的反应物,你的研究也会进行地很顺利,但是笔者还是建议你在做一个新合成反应时也要自己亲自试试不按照文献里建议的反应条件会发生什么结果。
参考文献
- [1] Pivsa-Art, S.; Satoh, T.; Kawamura, Y.; Miura, M.; Nomura, M. Bull. Chem. Soc. Jpn., 1998, 71, 467. DO: 10.1246/bcsj.71.467
- [2] (a)Ueda, K.; Yanagisawa, S.; Yamaguchi, J.; Itami, K. Angew. Chem., Int. Ed. 2010, 49, 8946. DOI:10.1002/anie.201005082 (b) Kirchberg, S.; Tani, S.; Ueda, K.; Yamaguchi, J.; Studer, A.; Itami, K. Angew. Chem., Int. Ed. 2011, 50, 2387. DOI:10.1002/anie.201007060
- [3] Colletto, C.; Islam, S.; Jukiá-Hernández, F.; Larrosa, I. J. Am. Chem. Soc. 2016, 138, 1677. DOI: 10.1021/jacs.5b12242
- [4] Ackermann, L.; Vicente, R.; Althammer, A. Org. Lett. 2008, 10, 2299–2302. DOI: 10.1021/ol800773x
- [5] Punji, B.; Song, W.; Shevchenko, G. A.; Ackermann, L. Chem. Eur. J. 2013, 19, 10605–10610. DOI: 10.1002/chem.201301409
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文投稿作者孙苏赟
平面对称在化学中是很常见的,一般来讲当一个化合物具有结构镜面对称并且性质相同的结构就称为面对称,当一个反应只在这两个结构中的一个上发生时就是去对称化。在有机合成中,去对称化有着广泛的应用,可以用作合成一些复杂的天然产物分子1,2,2016年Inoue组合成的ryanodine就是一个例子。

SCHEME1|a)去对称化的目标导向性合成的分子;b)使用二环[4.4.1]三酮作为重要中间体合成actinophyllicacid(1)的前体
不久前,首尔国立大学David Y.-K.Chen课题组和北海道大学的Hidetoshi Tokuyama课题组联合在ACIE上报道了利用以上去对称化的思路进行的antinophyllicacid的全合成工作,尽管这个分子在之前有被其他课题组合成过3-7,但是小编觉得本文报道的方法更加巧妙。
“Total Synthesis of Actinophyllic Acid
Yu Yoshii, Hidetoshi Tokuyama, and David Y.-K. Chen*
Angew. Chem. Int. Ed. 2017, 56, 12277-12281 DOI: 10.1002/anie.201706312

SCHEME2|Actinophyllicacid盐酸盐的合成(1·HCl).
合成从二环[4.4.1]三烯酮出发,先是用TsOH和乙二醇保护羰基,再小心控制反应条件,NMO/OsO4进行不对称双羟基化得到3,可喜的是这个过程并不会影响旁边两个共轭的双键。之后再用Pb(OAc)4进行邻二醇的裂解得到二醛中间体,再用NaBH(OAc)3还原、PMBNH2成环得到的4用TeocCl换上保护Teoc的化合物5。这里将PMB换成Teoc是因为EWG会使分子在接下来的氧化反应等反应中收到尽可能少的影响。

SCHEME3|a)利用去对称话策略对5的处理的早期研究;b)合成不对称烯烃17和二硅醚化合物20,21
当得到了5之后就要进行关键的步骤了(Scheme3),如果从目标分子actinophyllicacid的角度来看,如何构造吲哚结构片段是很重要的,所以在scheme3a中是一些进行测试的可能可行的方案。氧化或是胺化条件的效果都不太好,但是后来发现用催化氢化的条件可以得到比例近似1:1的17和18,但是生成18的机理尚不是很清楚8。尽管如此,18在双羟基化的条件下(OsO4,[K3Fe(CN)6],58%yield)可以得到二醇19,再利用TBSCl进行去对称化得到硅醚化合物20,再利用IBX进行氧化即得到了重要的中间体21。这样的合成策略在合成中是非常有用的。
接下来来看通过中间体17,20和21合成actinophyllicacid的途径,特别是构造其中的吲哚结构(此前也有人对此进行研究过4-7)。基于以上的研究,回到scheme2的合成路线中,5和6进行脱溴的[2+3]环加成反应原位得到异恶唑啉化合物7,产率68%,并且没有异构体产生。溴代异恶唑啉7之后在MeOLi作用下脱溴,DDQ氧化得到8,8是一个缩酮类,并且在C2的氧化态是符合目标分子的要求的。

SCHEME4|偶联反应的筛选条件
选择性的将2中三个双键中的两个进行官能团化,多步处理后得到了还有剩余一个双键的8,8接下来就是要进行芳构化反应以进行后续的吲哚结构的构造;此外,通过将其与邻吡咯烷进行同步化,可以使得芳基化更容易发生;这里利用的是Wolfe开发的方法学,反应中可以使双键的两个碳原子的同时偶联反应10。将8用TFA处理出去二胺的保护剂形成TFA的盐,而这个中间体就可以直接用芳胺化反应条件进行处理了。10的产率是和反应条件息息相关的(Scheme4),找寻到的最佳反应条件是p-NO2BrPh,Pd(OAc)2,P(2-furyl),Cs2CO3和THF作为溶剂。

SCHEME5|胺化芳化过程可能的机理的推测
通过二维NMR和一系列研究最终确定了10的成功合成。可能的反应过程是初始产物发生异构化,或者是芳化反应的发生可能和氨基无关。但是后来用同位素标记法的试验结果并不支持这样的推测(Scheme5a,b)。但是后来发现脱胺保护基之后会生成化合物22(Scheme5c),但是这个结构并不能在一般的条件下进行接下来的芳化反应(Scheme5d)。因此作者提出了Scheme5e中的模型。
再回到scheme2中,以上的胺化芳化的方法可以将8转化成的10,10在酸性条件下脱去保护剂得到11,之后可以自发的发生反应生成具有吲哚片段的化合物12,12在羧基的α-位增碳成环最终得到目标分子actinophyllicacid。
在上述的合成中,从二环化合物2出发,利用一系列方法学,包括非常新颖的双键双位偶联反应,这个步骤在以往对天然产物的合成中是非常少见的。
参考文献
- The Logic of Chemical Synthesis, Wiley-VCH,Weinheim,1989
- Tetrahedron Lett. 2014, 55, 7147;DOI:1016/j.tetlet.2014.10.152
- J. Am. Chem. Soc. 2008,130, 7568; DOI:10.1021/ja803158y
- J. Am. Chem. Soc. 2010,132,4894; DOI:10.1021/ja100178u
- J. Am. Chem. Soc. 2013,135,12984; DOI:10.1021/ja4070206
- Tetrahedron, 2014, 70, 4094; DOI:1016/j.tet.2014.03.034
- J. Am. Chem. Soc. 2016,138, 3298; DOI:10.1021/jacs.6b00567
- J. Am. Chem. Soc.1987,109, 6115; DOI:10.1021/ja00254a034
- J. Org. Chem. 2008, 73, 8851; DOI:10.1021/jo801631v
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要

Somei-Kametani 反应(Somei-Kametani reaction)又称为Somei-Kametani偶联(Somei-Kametani coupling),是在三正丁基膦催化下,碳亲核试剂(尤其硝基烷烃)与芦竹碱(gramine又称为禾草碱)及其衍生物之间的选择性单烷基化过程。该反应由日本神奈川县立大学(Kanazawa University) 的染井正德(Somei Masanori)与龟谷哲治(Kametani Tetsuji)1981年首次报道。该反应现已广泛应用于各类天然产物的全合成。
基本文献
- [1] M. Somei, Y. Karasawa, C. Kaneko, Heterocycles, 1981, 16, 941. doi: 10.3987/R-1981-06-0941.
- [2] T. Kametani, N. Kanaya, M. Ihara, J. Chem. Soc., Perkin Trans. 1, 1981, 959. doi: 10.1039/P19810000959.
反应机理

反应实例
epi-notoamide E 的全合成[1]

硝基烷烃参与的单烷基化[2]

clavicipitic Acid的形式全合成[3]

具有季碳中心的thioindoline的非对映选择性合成[4]

实验步骤
向三口烧瓶中加入芦竹碱(1 eq.)与硝基烷烃(3.5 eq.)的乙腈溶液(0.3 M),开启搅拌。随后,再向上述反应混合物中加入n-Bu3P (0.3eq.)及适量乙腈溶剂。在氩气气氛下,将上述反应液回流4 h。减压除去溶剂后,将上述反应液用0.5M HC1酸化,并用DCM-MeOH (19:1 v/v)萃取,将合并的有机相依次用饱和食盐水洗涤及无水Na2SO4干燥,减压除去溶剂后,将所得油状物通过PTLC分离纯化,获得最终目标产物。
实验技巧
参考文献
- [1] T. J. McAfoos, S. Li, S. Tsukamoto, D. H. Sherman, R. M. Williams, Heterocycles. 2010, 82, 461. doi: 10.3987/COM-10-S(E)19.
- [2] M. Somei, Y. Karasawa, C. Kaneko, Heterocycles, 1981, 16, 941. doi: 10.3987/R-1981-06-0941.
- [3] D. A. Boyles, D. E.Nichols, J. Org. Chem. 1988, 53, 5128. doi: 10.1021/jo00256a039.
- [4] A. V. Novikov, A. Sabahi, A. M. Nyong, J. D. Rainier, Tetrahedron: Asymmetry 2003, 14, 911. doi: 10.1016/S0957-4166(03)00077-6.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
Robert J. P. Corriu(1934年-2016年2月 ) 法国化学家。
经历
1961 蒙彼利埃大学博士課程修了
1963 佩皮尼昂大学助教授
1964 普瓦捷大学准教授
1969 蒙彼利埃大学教授
获奖经历
1969 法国化学会赏(Prix Sue)
1982 法国国立科学研究中心(CNRS)银牌奖章
1984 ACS Kipping Award
1985 法国化学会赏(Prix Lebel)
1992 洪堡赏
1994 马克斯 普朗克赏
1998 Wacker・Silicone 赏
2005 Wittig-Grignard赏
研究概要
他毕生致力于金属有机化学领域、特别是以有机硅化合物、有机镁化合物、有机磷化合物、过渡金属催化剂、高配位硅及膦化合物为中心展开的研究。
- 熊田-玉尾-Corriu偶联反应的开发[1]
在镍或者钯催化剂的作用下,卤代芳烃和Grignard试剂反应,形成新的碳-碳键的反应叫做Kumada-Tamao-Corriu偶联反应。Corriu和京都大学的熊田/玉尾在同一时期各自独立开发了该反应。最近几年,这一反应逐渐成为人们熟悉的人名反应—-熊田-玉尾-Corriu偶联反应。该反应是最早的金属催化的sp2-sp2碳碳键构筑反应,后来把该类反应统称为偶联反应,Corriu和Kumada等人是致力于偶联反应最早的一批研究者,与偶联反应诺贝尔奖获得者有着可比肩齐的研究贡献。
- 由分子前体合成及评价陶瓷、纳米材料[2]
评论&其他
- 共著论文共有700多篇以上
相关动画
相关文献
- Corriu, R. J. P.; Massse, J. P. J. Chem. Soc., Chem. Commun. 1972, 144. DOI: 10.1039/C3972000144a
- Corriu, R. J. P, Angew. Chem. Int. Ed, 2000, 39, 1376. DOI: 10.1002/(SICI)1521-3773(20000417)39:8<1376::AID-ANIE1376>3.0.CO;2-S
- Mehdi, A.; Reye, C.; Corriu, R. Chem. Soc. Rev. 2011, 40, 563. DOI: 10.1039/B920516K
含有光氧化還原過程的雙催化反應
陽光是自然界中獨特的清潔能源,因為它廉價、無污染而且太陽無時無刻不在向外輸出光能。早在1912年,化學家Giacomo Ciamician就提出了僅使用太陽光就可以完成一個有機化學反應的設想。[1]然而,大多數有機化合物並不吸收可見光,這導致很多光化學反應都是由紫外光完成的。[2]直到2008年,普林斯頓大學的D. W. C. MacMillan以及威斯康辛大學的T. P. Yoon分別發現某些光敏劑在吸收可見光后形成相應的激發態物種通過單電子轉移 (SET) 過程活化有機物。這種新的方法在近五年得到了很大的發展。[3]
美中不足的是大多數利用可見光氧化還原催化的反應後續過程都類似於自由基反應,這也就決定了這類反應只能在“產生自由基”與尋找“自由基接受體”這兩方面做文章,極大的限制了反應底物的選擇。2011年致力於Pd催化研究的Sanford教授,利用中心金屬可以與自由基結合形成高一個氧化態的金屬配合物這一特點,完成了photoredox/ palladium共同催化的芳基化反應。這也是首例可見光-過渡金屬雙催化反應的報道。[4]光催化劑與金屬催化劑在同一個轉化中串聯運轉、各取所長,使兩種催化劑獨自不能實現的反應得到了實現,提供了化合物合成的全新的方法。此後,光催化劑 (PC) 與Au[5]、Cu[6]、Ru[7]、Co[8]等過渡金屬雙催化反應均有報導。

Figure 1 自由基與過渡金屬結合
光氧化還原與鎳催化劑通過單電子轉移實現2°烷基交叉偶聯
一項近期的統計表明,在藥物化學家完成的C-C鍵生成反應中,超過60%的反應可以歸類于過渡金屬催化的交叉偶聯。[9]這說明了偶聯反應對現代化學的發展極其重要,某些技術性的難題也亟待解決。目前,C(sp2)-C(sp2) 鍵偶聯的研究最為廣泛透徹。然而,C(sp2)-C(sp3)鍵的構建仍然是具有挑戰性的,很多方法並不能普遍的推廣。

Figure 2 Pd(0) 催化C-C键偶联机理 (摘自Science, 2014, 345, 433.)
以Pd(0)為代表的交叉偶聯反應大多數是基於雙電子轉移過程:氧化加成、轉金屬、還原消除。當烷基試劑作為親核體時,轉金屬化就成為了反應的決速步。為了加快轉金屬化過程,Grignard試劑參與的Kumada-Corriu反應以及有機鋅試劑參與的Negishi反應均有所研究[10]。然而這類金屬有機試劑並不穩定,它們對空氣與潮濕敏感,操作比較困難。由於該試劑有強親核性及鹼性,此類反應官能團兼容性低,反應副產物多,整體的反應效果不甚理想。烷基硼酸或烷基氟硼酸鹽也是一類烷基親核試劑。由於有機硼試劑性能溫和且絕大多數對空氣穩定易於存放,製備硼試劑的方法較烷基金屬試劑更多,因此Suzuki-Miyaura反應備受科研工作者青睞。可是C(sp3)-硼試劑轉金屬的方法的研究還僅僅停留在在初級階段。[11]由於其親核性較弱,為提高反應產率,經常使用2-4個當量的鹼以及很高的反應溫度(~150℃),甚至是等當量的銀鹽或銅鹽。[12]於此同時,Pd作為這類反應的催化劑也有一定的局限性,即:(1)需要使用高溫條件及過量的鹼,使反應總體效益降低;(2)儘管多種Pd催化劑、配體、條件篩選等工作已經嘗試,但是諸如2-甲基烷基硼試劑這樣的高取代的脂肪基無法得到相應的偶聯化合物。
隨著可見光-金屬雙催化領域的發展,Molander教授團隊希望通過新的單電子轉移(SET)過程實現C(sp2)-C(sp3)鍵的構築。實現這個想法的一個挑戰在於,通過光氧化形成自由基,所形成的自由基需要被中心金屬俘獲使金屬被氧化,這樣才能進行接下來的光還原。Akita組早前的工作證明了在光照下以Ir[dFCF3ppy]2(bpy)PF6為催化劑可以使2°烷基氟硼酸鉀產生相應的烷基自由基。[13]金屬催化劑,從與芳基鹵反應性強且具有單電子轉移能力的Ni鹽開始研究。經優化后得到如下條件。

Figure 3 Molander課題組的最優條件 (摘自Science, 2014, 345, 433.)
其反應機理如下圖所示:

Figure 4 可見光-Ni催化循環圖 (摘自Science, 2014, 345, 433.)
光催化與Ni催化協同進行。光氧化過程產生了2°烷基自由基,金屬試劑捕獲后發生還原消除,再通多對Ni(I)的光還原使得金屬催化劑再生。
有趣的是,在Molander小組發現的同時,普林斯頓大學MacMillan教授課題組也發現了類似的反應。[14]這兩篇背靠背的工作,發表于同一期的Science雜誌中。MacMillan組使用烷基羧酸作為烷基源,在研究過程中他們發現bench-stable的NiCl2·glyme的效果比Ni(COD)2更好。其催化循環機理類似Molander的工作,但他們推測Ni(0)是光催化劑與Ni(II)發生雙電子轉移形成的,過量的氨基酸還原了光催化劑,其機理如下:


Figure 5 MacMillan課題組推測反應機理 (摘自Science, 2014, 345, 437.)
這兩個類似的反應,僅需要在惰性氛圍下,使用家用燈泡在室溫下照射,加入1%-2%光催化劑與3%-10%的鎳催化劑,通過單電子轉移過程巧妙的迴避了傳統方法裡面烷基親核試劑轉金屬化慢的問題,避免了高溫以及更加昂貴的Pd催化劑的使用。Molander小組對此反應做了一個競爭實驗,將等量的芳基硼試劑與烷基硼試劑投入反應。結果高產率得到烷基硼試劑的偶聯產物,未得到相應的芳基產物。這是由於烷基硼試劑單電子轉移速率快的原因。在另一個機理實驗中,外消旋化的烷基硼試劑在不對稱配體的調控下,反應產物得到了立體富集,而Pd催化通常會立體保持。因此,這種方法可以很好地與之前已開發的催化方法互補。
此後,Molander小組通過理論計算驗證推測的機理。[15] 通過計算,他們發現Ni催化劑可能有兩種反應途徑。除了如上所述的過程,還有可能Ni(0)先捕獲自由基,接著氧化加成,再還原消除,最後催化劑再生。

Figure 6 以2,2′-連吡啶為配體進行理論計算結果及可能的反應途徑 (J. Am. Chem. Soc. 2015, 137, 4896.)
在進一步摸清可見光氧化還原-Ni雙催化體系的機理后,Molander課題組將其在多種類型2°烷基硼試劑進行了一系列的拓展,獲得了較好的產率,證明了此方法具有可觀的普適性。[16]-[19]

Figure 7 Molander課題組對雙催化烷基化反應的拓展
Molander教授很興奮的表示,這種方法極大的簡化了反應的操作也極大的推動了2°烷基交叉偶聯反應的發展。但唯一的缺點是,由於烷基自由基的產生,該方法可能使原本具有立體專一性的烷基外消旋化,如何通過配體調控解決這一問題將是該課題組以後工作的重點。
相关文献
- T. P. Yoon, M. A. Ischay, J. Du Nature Chem., 2010, 2, 527. DOI: 10.1038/nchem.687
- N. Hoffmann, Chem. Rev. 2008, 108, 1052. DOI: 10.1021/cr0680336
- C. K. Prier, D. A. Rankic, D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322. DOI: 10.1021/cr300503r
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt, M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566. DOI: 10.1021/ja208068w
- A. Tlahuext-Aca, M. N. Hopkinson, B. Sahoo, F. Glorius, Chem. Sci., 2016, 7, 89. DOI: 10.1039/C5SC02583D
- Y. Ye, M. S. Sanford J. Am. Chem. Soc., 2012, 134, 9034. DOI: 10.1021/ja301553c
- D. C. Fabry, M. A. Ronge, J. Zoller, M. Rueping, Angew. Chem. Int. Ed., 2015, 54, 2801. DOI: 10.1002/anie.201408891
- G. T. Zhang, C. Liu, H. Yi, Q. Y. Meng, C. L. Bian, H. Chen, J. X. Jian, L. Z. Wu, A. Lei, J. Am. Chem. Soc., 2015, 137, 9273. DOI: 10.1021/jacs.5b05665
- S. D. Roughley, A. M. Jordan J. Med. Chem., 2011, 54, 3451. DOI: 10.1021/jm200187y
- E. Jahn, U. Jahn, Angew. Chem. Int. Ed., 2014, 53, 13326. DOI: 10.1002/anie.201408748
- J. C. Tellis, D. N. Primer, G. A. Molander, Science, 2014, 345, 433. DOI: 10.1126/science.1253647
- (a) D. Imao, B. W. Glasspoole, V. S. Laberge, C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024; DOI: 10.1021/ja8094075 (b) J. Z. Deng, D. V. Paone, A. T. Ginnetti, H. Kurihara, S. D. Dreher, S. A. Weissman, S. R. Stauffer, C. S. Burgey, Org. Lett., 2009, 11, 345. DOI: 10.1021/ol802556f
- (a) Y. Yasu, T. Koike M. Akita* Adv. Syn. Cat., 2012, 354, 3414; DOI: 10.1002/adsc.201200588 (b) K. Miyazawa, Y. Yasu, T.Koike, M. Akita, Chem. Comm., 2013, 49, 7249. DOI: 10.1039/C3CC42695E
- Z. W. Zuo, D. T. Ahneman, L. L. Chu, J. A. Terrett, A. G. Doyle, D. W. C. MacMillan, Science, 2014, 345, 437. DOI: 10.1126/science.1255525
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander, M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896. DOI: 10.1021/ja513079r
- D. Ryu, D. N. Primer, J. C. Tellis, G. A. Molander, Chem. Eur. J., 2016, 22, 120. DOI: 10.1002/chem.201504079
- I. Karakaya, D. N. Primer, G. A. Molander, Org. Lett., 2015, 17, 3294. DOI: 10.1021/acs.orglett.5b01463
- M. E. Khatib, R. Augusto, M. Serafim, G. A. Molander, Angew. Chem. Int. Ed., 2016, 55, 254. DOI: 10.1002/anie.201506147
- D. N. Primer, I. Karakaya, J. C. Tellis, G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195. DOI: 10.1021/ja512946e
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
通常日本科学界的十月,最受关注的事情就是诺贝尔奖了吧,这一点我已经深深的感受了好几年。并且这已经是我第三度近距离感受日本诺贝尔奖效应了,现在成了科研加媒体工作者双重身份,对这样的科学大事件也就越来越多了一些见解和责任。先就说说这三次发生在身边的诺贝尔奖吧:
第一次是五年前,来日本冈山大学西原康师教授组读博的一个周后,十月初的一天2010年的诺贝尔化学奖揭晓了,铃木章(Akira Suzuki)、根岸英一(Eiichi Negishi)还有美国的理查德-赫克(Richard F. Heck)三位教授获奖了,终于有一次知道诺贝尔获奖者是做了些什么的了,说实话,那会儿哪里谈得上什么学术造诣,知道的关于诺贝尔奖的常识就是诺贝尔本人发明了炸药,还有居里夫人级别的伟人科学家们,真是对近现代的诺贝尔奖获得者没有概念。而这是第一次,因为硕士期间研究室做了很多绿色体系的Suzuki和Heck偶联,写论文的序言总有偶联反应的历史背景和意义等等,所以深有感受他们在这个领域的卓越成就,自己也算是对这个领域的研究小有涉足,所以自然多了一些自豪,而且根岸英一教授我在他未获奖之前还是见过本人的,硕士阶段来日本交流时在冈山听过他的报告,这一下让我觉得,哇塞,原来诺贝尔奖离我们这么近。

这才是小小的开场,之后让人感受强烈的诺贝尔奖效应来了,从表及里,从小范围到大范围。最先是整个研究室忙碌起来了,我博士期间导师西原教授因为和根岸英一老师的关系甚好,从“学缘”上也很紧密,自然也就成了这个效应最直接受影响的“当事人”之一。根岸老师常年居住在美国,与日本的联系最多的的就是和北海道大学的高桥保教授和冈山大学的西原老师,当时记得冈山的新闻媒体纷纷电话来道贺邀约,西原老师办公室的电话已经快被打爆了,叫了好几个学生去帮忙接电话、做记录,西原老师小小的办公室,也因为根岸老师的关系,一周内每天在外面等候的记者多的,前脚出去后脚进来也不过如此。半个多月,国内媒体大幅度报道这次诺奖,铺天盖地的各种新闻发布会、采访,报纸人物传记等等纷至沓来,两位老教授一下子成了国宝级人物。这些都是表象上最明显感受到的。大众媒体报纸毕竟不是解读科学领域的行家,偶尔看到一些节目不专业的用词和解释很好笑,挑剔的日本学生自己也都拿某些段子来吐槽和搞笑,(哈哈,即使是日本人也难免出错)在那一阵,确实是全民都在关注科学,连中学生也能说上一两句和诺奖有关的话题。
从更深层次上,2010年的诺奖对日本科学界的影响,让偶联反应成了日本最引以为傲的有机化学领域的成就,没有之一,并更加为世界瞩目,不光是这几个获了奖的偶联反应,其他一些同时期发现的金属偶联反应比如Hiyama,Kumada, Sonogashira等偶联反应尽管在名气上略逊一筹,也被大家放在一起去说。个人觉得,它们之间相互成就,相互补充完善,共同为这一系列的反应奠定了非常扎实的理论基础,极大推动了该研究在学术和工业领域的大力发展。
对我个人而言,这一次的诺奖效应绝对是对我科研生涯影响最大的一件事情,硕士阶段做水相中的偶联反应,比较偏实际应用的绿色化学,而在之后的三年博士阶段,也继续研究偶联反应在功能分子合成的应用上,接触了更多偶联反应还有不同体系的催化条件,可以说借着偶联反应的获奖,我和它的缘分继续,把偶联反应从简单实用做回了深入精细。
而对根岸老师,在日后也有了更多的了解,获奖之后他成了冈山大学的荣誉教授,每年基本都会来冈山一次,对他的代表研究,Negishi试剂,Negishi偶联反应,做研究时都接触过一段时间,颇为了解,近年来的ZACA反应,虽没深入了解,倒是在报告上听过几次。对根岸老师的印象,他的面像虽很和蔼,总是微笑,但却不免让人感觉到一种内在的严格,而且科学大家往往都不接地气,再加上年纪相差悬殊,和我们学生自然没有太多直接的语言和精神交流,唯独让我印象深刻的是他每次到各处去做邀请报告,都要带上他的夫人,是很和蔼的日本老太太,前几年老夫人腿脚还可以,能慢慢走,这两年慢慢腿脚不好需要坐轮椅,即使是这样,根岸老师去哪里也还是要带上她一起。之后有一次国际会议上,根岸老师“任性”的在会议晚宴上和钢琴手配合即兴演唱一首经典名曲,掌声四起,他就又接着唱,夫人则在远处的轮椅上安静的坐着,我因为见过她几面,就过去聊天,我说根岸“先生”(老师)唱的很好,老夫人小声念叨说:“唉,他就是喜欢唱歌,总是这样,很难停下来,真的好烦吧”我知道这是说给我们听的,琐碎的话语中看似抱怨却透着对他的包容。再后来看过Negishi获诺奖介绍的纪录片,他的女儿在纪录片中讲她的妈妈当年为了支持爸爸在美国的研究,在背后默默付出了很多很多,这样也就对他们的今天多了更多的理解和敬佩。
那几年从冈山西原研究室毕业的同学每人手里都有一份来自根岸老师的礼物,他的个人签名祝语一张,是西原老师为大家争取的福利之一,这个简单而珍贵的礼物至今都在我的桌子旁边,和西原研究室大家送我的临别祝语一样宝贝,根岸老师写给我们话也是我的座右铭之一,累了,挫败了,想放弃的时候看看

“大きな夢を持ち、追い続けよう!”(拥有大的梦想,并不断追寻下去)
上篇完。。。
根岸英一 (Eiichi Negishi,1935年7月14日-)是长居美国的日本有机化学家(照片:成功大学)。美国普渡大学Herbert C. Brown组特別教授。2010年因其在「钯催化的偶联反应」中的贡献获得诺贝尔化学奖。
经历
1958 东京大学 毕业
1958-1960 帝人株式会社
1960-1963 宾夕法尼亚大学 博士学位 (A. R. Day教授)
1963-1966 帝人株式会社
1966-1968 普渡 博士研究员 (Herbert C. Brown教授)
1968 普渡大学 助教授
1972 锡拉丘兹大学 助教授
1979 锡拉丘兹大学 教授
1979 普渡大学 教授
1999 普渡大学Herbert C. Brown特別教授
获奖经历
1960-1961 Fulbright-Smith-Mund Fellowship
1962-1963 Harrison Fellowship at University of Pennsylvania
1996 A. R. Day Award
1997 日本化学会奖
1998 Herbert N. McCoy Award
1998 ACS Award for Organometallic Chemistry
1998-2000 Alexander von Humboldt Senior Researcher Award
2000 Sir Edward Frankland Prize Lectureship
2003 Sigma Xi Award
2007 山田・古贺奖
2007 Gold Medal of Charles University
2010 ACS Award for Creative Work in Synthetic Organic Chemistry
2010 诺贝尔化学奖
2010 文化勋章・文化功劳者
研究概要

图片来自Org. Chem. Front., 2015, 2, 872–873
- 有机锌・有机铝・有机锆化合物的偶联反应的开发
根岸偶联反应这一被人熟知的人名反应已经广泛用于合成化学反应中,是学术研究及工业领域的合成化学家们最常用的反应之一。因偶联反应涉及许多领域,这一反应的开发受到科学界较高的评价,并因此根岸教授和其他两名化学家Heck、铃木章一同被授予2010年诺贝尔化学奖。

- 使用低原子价态的有机锆试剂的合成反应开发
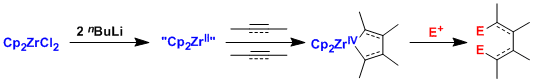

丁基锂和二氯二茂锆反应后得到的二价锆试剂,被称为根岸试剂,是可在各类不饱和键之间形成新的碳碳键的一种非常有效的试剂。
- 锆催化剂催化的铝羰基反应(ZACA)的开发和其不对称化反应。
评论&其他
- 是钯催化化学的发展历史上贡献卓越的科学家之一。
- 生于旧满洲国(伪满洲国)・新京(现:吉林省长春市)。(唯一一位生于旧满洲国的诺贝尔化学家,其余两位幼年时期在旧满洲国生活过的诺贝尔化学家还有白川英树,下村脩)
- 2010年是根岸夫妻的金婚之年。
- 诺贝尔化学奖获奖者名单公布期间,那个时候,他在电话旁边不停走来走去,犹豫等待着电话。
- 「劳逸结合,工作和游玩交替,是非常聪明的研究者」(之前所在的帝人公司的同事评价他)
- 卡拉OK的拿手曲目有1000曲之多。
- 执笔写过400篇以上的论文。
- 他在美国待了50年多年。家庭成员有妻、二女和四个孙子(女)。
- 2015年7月14日是他80岁生日,在他门下的众多研究者们纷纷前往学术研讨会及祝寿宴,许多媒体杂志也纷纷发表消息祝贺。
関連動画
相关文献
[1] Negishi, E.; Wang, G.; Rao, H.; Z. Xu. J. Org. Chem. 2010, 75, 3151. DOI: 10.1021/jo1003218
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!