作者:杉杉
导读:
近日,美国California Institute of Technology的G. C. Fu课题组在Angew. Chem. Int. Ed.中发表论文,报道一种全新的通过铁催化剂促进的烷基卤与烯基化合物之间的还原交叉偶联反应方法学,进而成功完成一系列烷基化合物的构建。
Iron-Catalyzed Reductive Cross-Coupling of Alkyl Electrophiles with Olefins
X.Tong, Z.Yang, C. Del Angel Aguilar, G. C. Fu, Angew. Chem. Int. Ed. 2023, ASAP. doi: 10.1002/anie.202306663.

正文:
近年来,通过铁催化剂促进的偶联反应方法学 (Figure 1A-B)[1]的相关研究已经逐渐受到有机合成化学家的关注。这里,受到近年来对于选择金属催化剂促进的烷基亲电底物与烯基化合物之间的还原偶联反应方法学 (Figure 1C)[2]-[3]相关研究报道的启发,美国California Institute of Technology的G. C. Fu课题组成功设计出一种全新的通过铁催化剂促进的烷基卤与烯基化合物之间的还原交叉偶联反应方法学 (Figure 1D)。

首先,作者采用1-碘-3-苯基丙烷与1-辛烯作为模型底物,进行相关反应条件的优化筛选 (Figure 2)。进而确定最佳的反应条件为:采用Fe(OAc)2作为催化剂,Xantphos作为配体,H-Si(OEt)3作为氢源,Mg(OEt)2作为添加剂,THF作为反应溶剂,反应温度为室温,最终获得相应的还原偶联产物1。

在上述的最佳反应条件下,作者分别对一系列烯基化合物以及烷基碘底物(Figure 3)的应用范围进行深入研究。


接下来,作者对上述还原交叉偶联过程的反应机理进行进一步研究 (Figure 4)。


同时,作者发现,采用Fe(OAc)2/Xantphos/Mg(OEt)2催化体系,能够将上述策略进一步应用于烯基化合物的加氢官能团化过程 (Figure 5)。

总结:美国California Institute of Technology的Gregory C. Fu课题组成功设计出一种全新的通过铁催化剂促进的烷基卤与烯基化合物之间的还原交叉偶联反应方法学,进而成功完成一系列烷基化合物的构建。这一全新的还原交叉偶联策略具有温和的反应条件、广泛的底物应用范围以及优良的官能团兼容性等优势。
参考文献:
- [1] A. Piontek, E. Bisz, M. Szostak, Angew. Chem. Int. Ed. 2018, 57, 11116. doi:10.1002/anie.201800364.
- [2] Y. Wang, N. C. Bruno, A. L. Placeres, S. Zhu, S. L. Buchwald, J. Am. Chem. Soc. 2015, 137, 10524. doi:10.1021/jacs.5b07061.
- [3] H. Pang, Y. Wang, F. Gallou, B. H. Lipshutz, J. Am. Chem. Soc. 2019, 141, 17117. doi:10.1021/jacs.9b04510.
- [4] M. Nakamura, K. Matsuo, S. Ito, E. Nakamura, J. Am. Chem. Soc. 2004, 126, 3686. doi:10.1021/ja049744t.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
作者:杉杉
导读:
近日,南开大学的徐大振与中国科学院大学的冯德鑫课题组在Green Chem.中发表论文,采用氧气作为氧化剂,首次报道一种全新的采用铁催化剂促进的分子内交叉脱氢偶联 (intramolecular cross-dehydrogenative coupling)反应(分子内C-H胺化)方法学,进而成功完成一系列咔唑与吲哚分子的构建。
Iron-Catalyzed Intramolecular C-H Amination for the Synthesis of N-H Carbazoles and Indoles
Z.Wang, Y.Zhang, J. Huang, J. Zhou, Y. Yu, D. Feng, D. Xu, Green Chem. 2023, ASAP. doi: 1039/D3GC00518F.

正文:
咔唑与吲哚结构单元广泛存在于各类天然产物以及生物活性分子中 (Figure 1)。并且,在过去的几十年里,构建咔唑与吲哚分子相关的合成转化策略研究, 一直备受有机合成化学家的广泛关注 (Scheme 1a)[1]-[5]。这里,受到近年来对于采用铁催化剂促进的通过需氧氧化 (aerobic oxidative)路径进行的C-H官能团化反应方法学[6]相关研究报道的启发,南开大学的徐大振与中国科学院大学的冯德鑫团队共同设计出首例通过铁催化剂促进的分子内交叉脱氢偶联 (分子内C-H胺化) 反应方法学 (Scheme 1b)。


首先,作者采用2-cyclohexenyl aminoarene 1a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用FeCl3作为催化剂, DMAc作为反应溶剂,反应温度为100 oC,最终获得95%收率的交叉脱氢偶联产物2a。

在上述的最佳反应条件下,作者分别对构建咔唑 (Scheme 2)以及吲哚(Scheme 3) 分子时的底物应用范围进行深入研究。




同时,该小组通过对于反应条件的进一步优化,将上述的分子内交叉脱氢偶联策略成功应用于3-芳基苯并呋喃衍生物6的构建 (Scheme 4)。

之后,该小组通过如下的一系列研究进一步表明,这一全新的交叉脱氢偶联策略具有潜在的合成应用价值 (Scheme 5)。

接下来,作者对上述偶联过程的反应机理进行进一步研究 (Scheme 6)。


基于上述的实验研究以及前期相关的文献报道[1]-[5],作者提出如下合理的反应机理 (Scheme 7)。

总结:南开大学的徐大振与中国科学院大学的冯德鑫团队共同设计出首例通过铁催化剂促进的分子内交叉脱氢偶联 (分子内C-H胺化)反应方法学,进而成功完成一系列咔唑与吲哚分子的构建。这一全新的分子内C-H胺化策略具有温和的反应条件、广泛的底物应用范围以及优良的官能团兼容性等优势。
参考文献:
- [1] W. C. P. Tsang, N. Zheng, S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560. doi: 10.1021/ja055353i.
- [2] S. Maity, N. Zheng, Angew. Chem. Int. Ed., 2012, 51, 9562. doi: 10.1002/anie.201205137.
- [3] A. Maity, B. L. Frey, N. D. Hoskinson, D. C. Powers, J. Am. Chem. Soc. 2020, 142, 4990. doi: 10.1021/jacs.9b13918.
- [4] R. Yu, D. Li, F. Zeng, J. Org. Chem. 2018, 83, 323.doi: 10.1021/acs.joc.8b02308.
- [5] X. Ning, X. Liang, K. Hu, C. Yao, J. Qu, Y. Kang, Adv. Synth. Catal. 2018, 360, 1590. doi: 10.1002/adsc.201701512.
- [6] R. Hu, D. Han, N. Li, J. Huang, Y. Feng, D. Xu, Angew. Chem. Int. Ed., 2020, 59, 3876.doi: 10.1002/anie.201913400.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
本文作者:杉杉
导读:
近日,德国Stuttgart大学的B. Plietker课题组在Angew. Chem. Int. Ed.中发表论文,报道一种全新的通过阳离子铁配合物促进的通过环丙基取代烯炔分子参与的环异构化反应方法学,进而成功完成一系列三环环丁烷分子的构建。
Iron-Catalyzed Cycloisomerization and C-C Bond Activation to Access Non-canonical Tricyclic Cyclobutanes
F. Kramm, F. Ullwer, B. Klinnert, M. Zheng, B. Plietker, Angew. Chem. Int. Ed. 2022, ASAP. doi: 10.1002/anie.202205169.

正文:
环异构化反应方法学是由简单分子作为起始原料,构建复杂环状分子的有效策略[1]-[3]。这里,受到近年来对于Fe催化的环异构化[4]-[5],Rh催化的[5+2]环加成[6]以及Au催化的环异构化[7]反应方法学相关研究报道的启发,德国Stuttgart大学的B. Plietker课题组成功设计出一种全新的通过阳离子铁配合物促进的通过环丙基取代烯炔分子参与的环异构化反应方法学 (Scheme 1)。

首先,作者采用环丙基烯炔衍生物12作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用[(Ph3P)2Fe(NO)(CO)]BF4(2)作为阳离子铁催化剂,DCM作为反应溶剂,反应温度为50 oC,最终获得80%收率的环异构化产物13。

在上述的最佳反应条件下,作者对一系列环丙基取代烯炔底物 (Scheme 2)的应用范围进行深入研究。


同时,该小组进一步将上述的标准反应体系应用于各类炔丙基烷基碳酸酯底物的脱羧环异构化 (Scheme 4)。

接下来,该小组通过一系列控制实验 (Scheme 5)的相关研究表明,炔丙芳基结构单元 (烯炔底物)中的+M取代基能够促进环异构化过程的进行;芳基环丙烷结构单元中的+M取代基能够降低上述环异构化过程中的原料转化率。

基于上述的控制实验研究,作者提出如下合理的反应机理 (Figure 1)。

总结:
德国Stuttgart大学的B. Plietker课题组成功设计出一种全新的通过阳离子铁配合物促进的通过环丙基取代烯炔分子参与的环异构化反应方法学,进而成功完成一系列三环环丁烷分子的构建。这一全新的环异构化策略具有温和的反应条件、广泛的底物应用范围以及优良的官能团兼容性等优势。
参考文献:
- [1] (a) P. A. Wender, A. G. Correa, Y. Sato, R. Sun, J. Am. Chem. Soc. 2000, 122, 7815. doi: 10.1021/ja001483+.
- (b) F. Inagaki, K. Sugikubo, Y. Miyashita, C. Mukai, Angew. Chem. Int. Ed. 2010, 49, 2206. doi:10.1002/anie.200906994.
- [2] (a) T. Torigoe, T. Ohmura, M. Suginome, Angew. Chem. Int. Ed. 2017, 56, 14272. doi: 10.1002/anie.201708578.
- (b) D. F. Fernández, C. A. B. Rodrigues, M. Calvelo, M. Gulías, J. Mascareñas, F. López, ACS Catal. 2018, 8, 7397. doi:10.1021/acscatal.8b02139.
- [3] (a) E. Jiménez-Núñez, C. K. Claverie, C. Nieto-Oberhuber, A. M. Echavarren, Angew. Chem. Int. Ed. 2006, 45, 5452.doi: 10.1002/anie.200601575.
- (b) S. Ferrer, A. M. Echavarren, Org. Lett. 2018, 20, 5784. doi: 10.1021/acs.orglett.8b02478.
- [4] (a) F. M. Miloserdov, M. S. Kirillova, M. E. Muratore, A. M. Echavarren, J. Am. Chem Soc. 2018, 140, 5393. doi:10.1021/jacs.7b13484.
- (b) X. Cheng, Z. Wang, C. D. Quintanilla, L. Zhang, J. Am. Chem. Soc. 2019, 141, 3787. doi: 10.1021/jacs.8b12833.
- [5] (a) J. Teske, B. Plietker, ACS Catal. 2016, 6, 7148. doi: 10.1021/acscatal.6b02260.
- (b) J. Teske, B. Plietker, Org. Lett. 2018, 20, 2257. doi: 10.1021/acs.orglett.8b00612.
- [6] P. A. Wender, H. Takahashi, B. Witulski, J. Am. Chem. Soc. 1995, 117, 4720. doi: 10.1021/ja00121a036.
- [7] (a) E. Jiménez-Núñez, C. K. Claverie, C. Nieto-Oberhuber, A. M. Echavarren, Angew. Chem. Int. Ed. 2006, 45, 5452. doi: 10.1002/anie.200601575.
- (b) S. Ferrer, A. M. Echavarren, Org. Lett. 2018, 20, 5784. doi: 10.1021/acs.orglett.8b02478.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
本文作者:杉杉
导读
近日,南开大学的徐大振课题组在Green Chem.中发表论文,报道一种采用具有二级C(sp3)-H键的相关底物参与的脱氢交叉偶联反应方法学,进而成功实现相应全碳四级中心的构建。这一策略中,采用FeCl2•4H2O作为催化剂,空气中的氧气作为终端氧化剂,能够在较为温和、无配体以及无碱存在的反应条件下,顺利实现各类吲哚-2-酮与吲哚之间的交叉偶联过程,并以优良的化学选择性,获得一系列具有全碳四级中心的3,3-二取代羟吲哚衍生物。
Iron-catalyzed oxidative bis-arylation of indolin-2-ones for direct construction of quaternary carbons
K. Wu, Y. Xu, L. Cheng, R. Wu, P. Liu, D. Xu, Green Chem. 2021 ASAP. doi: 10.1039/D1GC02886C.

正文
过渡金属催化的交叉偶联反应方法学是当代有机合成化学中构建C-C键的有效策略之一。其中,在氧化条件下,实现两种不同类型的C-H键之间的交叉偶联过程,已经开始受到诸多研究团队的关注[1]。然而,这一类型的氧化CDC策略中,需要选择化学计量的氧化剂,形成反应过程中较为关键的自由基中间体,进而极大降低这一转化过程中的原子经济性。同时,在大规模的工业合成中,采用上述的氧化CDC策略,容易产生较为严重的安全问题。因此,仍需要开发一种更为绿色并且环境友好的C-C键构建的全新策略。
由于四重碳取代基之间存在高度的立体位阻,因此,通过交叉偶联策略构建全碳四级中心的相关研究仍面临巨大挑战[2]。并且,在目前已经报道的各类CDC策略 (Scheme 1a)[3]中,主要存在如下弊端:(1) 需要采用化学计量的各类化学氧化剂 (2) 采用有毒有害的有机溶剂 (3)需要强碱的参与,并涉及较为苛刻的反应条件。为解决上述策略中存在的诸多弊端,作者开始致力于设计全新的合成转化策略,进而能够在无碱存在的反应条件下,选择空气作为氧化剂,并采用一系列具有二级C(sp3)-H键的有机底物,进而顺利实现全碳四级中心的构建。然而,通过具有二级C(sp3)-H键的有机底物,化学选择性地构建全碳四级中心,同样面临如下挑战:(1) 与三级碳自由基相比,二级碳自由基的形成更为困难,并缺乏一定程度的稳定性 (2) 在涉及不同自由基中间体参与的合成转化过程中,反应体系可能十分复杂,并存在诸多不同类型的副反应,进而形成一系列不同形式的副产物。
同时,羟吲哚结构单元广泛存在于一系列天然产物以及合成药物分子中。尤其具有全碳四级中心的3,3-二取代羟吲哚为构成一系列生物碱类天然产物与非天然活性分子的重要结构单元 (Fig. 1)。这里,受到本课题组前期研究报道[4]的启发,南开大学的徐大振课题组报道首例铁催化的交叉脱氢偶联反应方法学,进而成功实现一系列3,3-二取代羟吲哚衍生物的合成 (Scheme 1b)。


首先,作者采用吲哚-2-酮1a与吲哚2a作为模型底物,进行相关偶联反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用FeCl2•4H2O作为催化剂,空气中的氧气作为终端氧化剂,在乙醇溶剂中,反应温度为65oC,最终获得93%收率的交叉脱氢偶联产物3a。

在上述的最佳反应条件下,作者首先对相应吲哚-2-酮底物的应用范围进行考察 (Table 2)。研究表明,一系列具有不同基团取代的吲哚-2-酮底物1均能够较好地与上述的标准反应条件良好地兼容,并获得相应的交叉脱氢偶联产物3a–3n (53-93% 反应收率)。同时,研究发现,C-5位具有供电子与吸电子基团取代的吲哚-2-酮底物,同样能够以较高的反应收率,获得相应的二芳基化产物3a–3g。之后,该小组进一步观察到,在芳基不同位置具有氯基团取代以及不同类型N-保护基团取代的吲哚-2-酮底物均能够有效地参与上述的交叉脱氢偶联过程,并获得中等至优良的反应收率 (3l–3n)。

接下来,作者进一步对一系列吲哚底物的应用范围进行考察 (Table 3)。研究表明,芳基中具有不同基团取代的吲哚底物,均能较好地与上述的最佳反应条件进行兼容,并获得相应的二芳基化产物3o–3z (76-93% 反应收率)。尤其对于具有强吸电子基团硝基取代的吲哚底物,同样能够表现出较为优良的官能团兼容性 (3u)。同时,上述的标准反应条件对于1-与2-位取代的吲哚以及3-甲基吲哚底物,同样能够有效地兼容,并获得相应的二芳基化产物3aa–3ac (80-92% 反应收率)。接下来,该小组进一步观察到,其它不同位置取代的吲哚底物,同样能够顺利参与上述的CDC反应过程,并获得相应的目标产物3ad–3ah (63-85% 反应收率)。

之后,作者对这一全新的CDC反应方法学的合成实用性进行研究 (Scheme 2)。该小组发现,将底物的用量扩大至4 mmol时,能够顺利完成海洋天然产物3a (1.16 g, 80% 反应收率)以及spermicidal 化合物3s的制备 (1.37 g, 81% 反应收率)。

接下来,为提出合理的反应机理,作者进行一系列相关的控制实验研究 (Scheme 3)。首先,作者发现,在1a与2a的标准反应体系中,加入2 eq. 自由基捕获剂时,反应过程受到较为显著的抑制。进而表明这一全新的CDC反应过程中涉及自由基中间体的形成 (Scheme 3a)。之后,该小组发现,将上述的CDC过程在在氩气气氛中进行时,则无法获得相应的二芳基化产物3a。这一实验观察表明,反应过程中,空气 (分子氧)的作用尤为关键 (Scheme 3b)。同时,作者进一步发现,在氩气气氛中,采用其它类型的氧化剂 (例如DTBP与TBHP)或将上述反应过程在200 mol% FeCl3•6H2O存在的条件下进行时,则仅能够获得痕量的偶联产物3a。这一事实表明,上述的合成转化过程中可能涉及含氧中间体的参与 (Schemes 3c-3d)。

基于上述的实验研究,作者提出一种合理的反应机理 (Scheme 4)。首先,通过螯合型Fe配合物M与1a之间的SET过程,形成相应的自由基中间体4。之后,通过自由基中间体4与氧气之间的偶联过程,形成过氧基中间体5。接下来,通过中间体5攫取1a中的氢原子,形成自由基中间体4与过氧化物6。其中,通过过氧化物6进一步失去羟基之后,再与Fe(III)进行配位,形成中间体7。再通过吲哚对于中间体7的亲核进攻以及后续的消除过程,形成化合物8。最终,通过化合物8的氧化脱氢过程,形成中间体9,并进一步与另一分子的吲哚底物作用,最终获得相应的目标产物3a。

总结
南开大学徐大振课题组成功设计出一种通过具有二级C(sp3)-H键的有机底物构建全碳四级中心的CDC反应策略。这一全新的CDC策略中,采用铁(II)盐FeCl2•4H2O作为催化剂,空气中的氧气作为终端氧化剂。同时,这一全新的CDC策略具有反应条件温和,优良的化学选择性,无需相应的配体以及碱的参与等优势。并且,这一全新的CDC方法学为一系列具有全碳四级中心的3,3-二取代羟吲哚分子的构建,开辟出一条更为简洁有效的反应途径。
参考文献
[1] (a) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh, A. Lei, Chem. Rev. 2017, 117, 9016. doi: 10.1021/acs.chemrev.6b00620.(b) M. S. Segundo, A. Correa, Synthesis, 2018, 2853. doi: 10.1055/s-0037-1610073.
[2] (a) K. W. Quasdorf, L. E. Overman, Nature, 2014, 516, 181. doi: 10.1038/nature14007.(b) H. Xie, J. Guo, Y. Wang, K. Wang, P. Guo, P. Su, X. Wang, X. Shu, J. Am. Chem. Soc. 2020, 142, 16787. doi: 10.1021/jacs.0c07492.
[3] (a) J. E. M. N. Klein, A. Perry, D. S. Pugh, R. J. K. Taylor, Org. Lett. 2010, 12, 3446. doi: 10.1021/ol1012668.(b) J. Wang, Y. Yuan, R. Xiong, D. Zhang-Negrerie, Y. Du, K. Zhao, Org. Lett. 2012, 14, 2210. doi: 10.1021/ol300418h.
[4] (a) R. Hu, D. Han, N. Li, J. Huang, Y. Feng, D. Xu, Angew. Chem. 2020, 59, 3876. doi: 10.1002/anie.201913400.(b) Y. Lai, R. Wu, J. Huang, J. Huang, D. Xu, Org. Lett. 2020, 22, 3825. doi: 10.1021/acs.orglett.0c01066.
(c) Z. Tan, K. Wu, L. Huang, R Wu, Z. Du, D. Xu, Green Chem. 2020, 22, 332. doi: 10.1039/C9GC03639C.
(d) L. Huang, D. Han, D. Xu, Adv. Synth. Catal. 2019, 361, 4016. doi: 10.1002/adsc.201900400.
(e) R. Hu, Y. Lai, D. Xu, Synlett 2020, 1753. doi: 10.1055/s-0040-1707195.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
导语
脂肪醇的选择性催化碳氢键官能团可以将广泛存在的醇类化合物转化为具有高附加值的复杂结构分子,因此该类转化也引起了化学家们的关注。通过氧自由基的生成以及1,5-氢迁移实现醇类化合物的远端碳氢键官能团化,是其中很经典的方法。常用的策略是将醇类化合物转化成为O-N化合物、高价卤、过氧化物、烷氧基铅等,以实现醇的“活化”促进氧自由基的生成。苏州大学朱晨教授、南开大学陈弓教授、北京大学焦宁教授、(从上海科技大学迁入)上海有机所左智伟教授等人通过使用Ir、Ru、Ag、Ce等催化剂促进游离醇生成氧自由基及1,5-氢迁移,实现了游离醇的远端碳氢键官能团化转化。如何使用廉价金属作催化剂仍然是一个挑战。近日,西安交通大学曾荣课题组报道了一种光诱导铁催化的新方法,脂肪醇在铁的催化下生成氧自由基、随后1,5-氢迁移、官能团化,进而实现了广泛易得的脂肪醇的远端胺化反应。

图1. 游离脂肪醇的远程官能团概念
Iron-Catalyzed Photoinduced Remote C(sp3)─H Amination of Free Alcohols.
Ni Xiong, Yang Li*, and Rong Zeng*
Organic Letters ASAP DOI: 10.1021/acs.orglett.1c03488
正文
近年来,西安交通大学曾荣教授课题组围绕光诱导铁催化反应展开了一系列研究工作。该课题组前期曾开发了光诱导铁催化下二级或三级醇的温和氧化制备芳香酸(Org. Lett. 2021, 23, 2915-2920)、烷基苯、聚苯乙烯氧化制备苯甲酸(Chin. J. Chem. 2021, 39, 3225-3230)等。基于对前期研究工作的深入探索,近日,该团队又报道了一种通过光诱导铁催化游离脂肪醇的选择性碳氢键胺化反应来构建复杂分子的新方法。

图2. 铁光催化游离脂肪醇的远程胺化(图片来源: Org. Lett.)
该反应具有良好的步骤、原子经济性及广泛的底物范围。在可见光促进铁催化下,不同的游离脂肪醇都可以实现远端胺化反应,高效、高选择性地得到相应的氨基醇类衍生物(图3)。

图3. 脂肪醇底物范围(图片来源: Org. Lett.)
此外,不同的偶氮类化合物也可以参与反应,得到相应的氨基醇类产物(图4)。

图4. 偶氮底物范围(图片来源: Org. Lett.)
通过简单的反应机理探索,作者也提出了该反应的催化循环(图5)。该反应由三价铁催化剂 Fe(III)和脂肪醇之间的光诱导电荷转移来引发,通过形成Fe(II)物种促使氧自由基中间体 Int-O的生成。分子内 1,5-氢转移(HAT)、接着与DBAD发生自由基加成产生氮自由基中间体 Int-N。最后,单电子转移生成胺化产物的生成以及再生Fe(III)催化剂。比较遗憾的是,对于氧自由基的生成机制,是通过氯自由基还是LMCT的过程,作者并没有给出结论。该实验室仍在进行更深入的机理研究。

图5. 催化循环(图片来源: Org. Lett.)
最后,作者还通过克级放大反应、麻氧化反应、Appel反应以及TsCl保护反应等进行了探索(图6),初步展示出了反应的实用性,也为后续的转化提供了基础。

图6. 产物衍生化(图片来源: Org. Lett.)
总结
西安交通大学曾荣教授课题组开发了一种高效的铁基光氧化还原催化系统实现了游离脂肪醇的远程C-H键胺化。该反应使用廉价易得的铁催化剂(FeCl3),结合可见光催化,具有良好的底物耐受性、条件温和、绿色经济和区域选择性良好等的优点。该研究成果以“Iron-Catalyzed Photoinduced Remote C(sp3)─H Amination of Free Alcohols”为题,发表于Organic Letters。西安交通大学化学学院博士研究生熊泥为该论文的第一作者,曾荣教授和李洋教授为本工作的通讯作者。
曾荣教授简介

西安交通大学化学学院教授,海外高层次人才计划、西交“青年拔尖人才计划A类”入选者,博导。2008年于浙江大学取得学士学位后直博本校,并于2013年取得博士学位,师从我国著名有机化学家麻生明院士,主要研究联烯参与的自由基加成以及碳氢官能团化。2013年至2016年,在美国德克萨斯大学奥斯丁分校从事博士后研究工作,合作导师董广彬教授,研究方向为非张力环酮的碳碳键活化偶联。2016年至2018年,在以色列威兹曼科学研究所从事博士后研究工作,合作导师David Milstein教授,研究方向为Pincer络合物合成以及在一氧化二氮活化中的应用。2018年起,在西安交通大学从事独立研究工作。目前在J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、ACS Catal.等国际主流学术期刊发表系列论文,授权中国专利3项。
Email: rongzeng@xjtu.edu.cn.
曾荣教授课题组简介:

曾荣教授课题组成立以来主要研究方向为基于协同催化策略的有机合成新方法研究,主要包含惰性键活化转化、光催化、活性杂环化合物合成、有机金属络合物的合成及应用等。
曾荣课题组欢迎硕士研究生、博士研究生的积极报考。Email: rongzeng@xjtu.edu.cn.
此外,因发展需要,曾荣课题组面向社会公开招聘青秀人才以及科研助理。
青秀人才个人待遇:享受学校提供青秀人才的一切待遇。详情请见:其中,A类青秀年薪30万;B类青秀年薪16万;此外,课题组奖励补助3-5万。(青秀A最高年薪35万)
青秀计划表现优异者,优先推荐申请西安交通大学青年拔尖人才计划。
研究助理:硕士毕业或即将毕业;具有有机化学背景;发表过英文论文,有一定的英文书写能力。表现优异者优先推荐博士申请。
李洋副教授简介

西安交通大学化学学院副教授,博士生导师。2010年于兰州大学取得学士学位。2015年于上海有机化学研究所取得有机化学博士学位,师从我国著名有机化学家丁奎岭院士,主要从事手性配体的设计合成以及不对称催化氢化反应的研究。2017年至2018年在美国斯克里普斯研究所做访问学者研究,合作导师余金权(Jin-Quan Yu)教授,研究方向为钯催化C-H键官能团化反应。2015年7月加入西安交通大学从事独立研究工作。主要从事过渡金属催化的惰性化学键活化及烯炔的官能团化反应研究。迄今为止国际主流学术期刊Angew. Chem. Int. Ed., Org. Lett., Org. Chem. Front., Chem. Eur. J.等发表论文多篇。
欢迎硕士研究生、博士研究生的积极报考。
Email: yanglee@mail.xjtu.edu.cn.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
本文作者:Joy
导读
浙江大学的陆展团队首次报道一种采用铁催化剂促进的由1,1-二取代烯基化合物参与的高度对映选择性氢化反应方法学,进而以良好的反应收率与优良的对映选择性,获得一系列手性烷基衍生物。首先,该课题组设计并合成出全新的手性8-噁唑啉亚氨基喹啉配体 (chiral 8-oxazoline iminoquinoline ligand, 8-OIQ)及其相应的铁配合物。之后,在上述手性铁配合物存在的条件下,作者发现,采用1 atm的氢气压力,即可完成相应的不对称氢化过程。这一全新的对映选择性氢化策略具有实验操作简洁以及良好的官能团兼容性等优势。此外,该小组通过氘标记实验,对相关的反应机理进行初步研究。
Iron-Catalyzed Highly Enantioselective Hydrogenation of Alkenes
P. Lu, X. Ren, H. Xu, D. Lu, Y. Sun, Z. Lu, J. Am. Chem. Soc. 2021, 143, 12433. doi: 10.1021/jacs.1c04773.

正文
金属催化的烯基化合物对映选择性氢化策略作为制备光学活性化合物的一种有效方法,在学术界与工业界中已经获得广泛的应用[1]。值得注意的是,这一策略中,主要采用具有手性膦配体配位的贵金属催化剂 (Scheme 1a) [2]。然而,由于贵金属在地球中含量的稀缺性,因此,合成化学家更多开始关注地球中含量较为丰富的金属在烯基衍生物催化不对称氢化反应中的应用[3]。铁作为地球中含量最丰富的过渡金属,不仅价格低廉,并且具有良好的生物相容性以及环境友好的优势[4]。然而,由于缺乏适宜的手性配体,因此,在铁催化的烯基衍生物催化不对称氢化反应方法学的研究中,将存在极大的挑战。近期,尽管已经开发出一系列能够应用于烯基化合物催化氢化方法学的膦基或吡啶基铁配合物 (phosphine- or pyridine-based iron complexes)[5],然而,采用铁催化剂参与的烯基衍生物的高度对映选择性氢化反应方法学,一直以来,仍未有相关的研究报道。而寻找或设计有效的手性配体或适宜的催化体系,是解决上述问题的关键。这里,陆展教授课题组成功设计出一种喹啉基N,N,N-三齿手性配体 (quinoline-based N,N,N-tridentate chiral ligand),进而将其成功应用于铁催化的1,1-二取代烯基化合物的高度对映选择性氢化反应策略的研究 (Scheme 1b)。

(图片来源:J. Am. Chem. Soc.)
最初,作者已经筛选出能够应用于铁催化烯基化合物不对称氢化过程的噁唑啉亚胺吡啶 (oxazoline minopyridine, OIP)铁配合物[6]。然而,却存在如下缺陷:(1) 反应过程中,能够进行异构化的副反应,形成难以去除的三取代烯基化合物;(2) 仅能够获得中等程度的对映选择性;(3)反应过程的重现性较差。为解决上述问题,作者进一步设计出一种全新的8-噁唑啉亚氨基喹啉 (8-oxazoline iminoquinoline, 8-OIQ)配体,其中选择喹啉骨架代替原配体中相应的吡啶结构单元 (Scheme 2)。

(图片来源:J. Am. Chem. Soc.)
之后,作者采用1,1-二取代烯基化合物1a与氢气之间的反应作为模型反应。进行相关反应条件的优化研究 (entry 1-11)。进而确定最佳的反应条件为:在室温、1atm氢气压力条件下,采用R、R’均为异丙基取代的手性Le·FeCl2配合物作为催化剂,选择CH3CN与n-C18H37SiH3作为添加剂,NaBHEt3作为活化剂,甲苯作为反应溶剂,反应时间为12 h。

(图片来源:J. Am. Chem. Soc.)
在上述最佳的反应条件下,作者对烯基化合物的底物应用范围进行研究 (Scheme 3)。作者发现,烯基化合物苯环中具有的富电子与缺电子取代基,例如醚键、硅醚、烷基、氨基、Bpin、硫醚以及卤素取代基,均能够与上述的最佳反应条件有效地兼容,并以优良的对映选择性获得相应产物2a–2m (> 92% ee)。并且,由于立体效应的存在,具有邻甲氧基苯基取代的烯基底物在10 mol % 手性催化剂存在下,尽管氢化过程够顺利进行,然而却获得具有较低 ee值的产物2n。同时,具有多环与杂环取代的烯基化合物 (1o–1q),同样能够良好地与上述的标准反应条件兼容。而且,具有脂肪族线性与支链 (1r–1x)以及苯基 (1y)取代的烯基底物,在上述的标准反应条件下,同样能够较为容易地转化为相应的手性产物 (88-98 % ee)。之后作者观察到,烷基链中具有氨基 (1z)与缩醛 (1aa, 1ab) 官能团取代的烯基底物,同样能够获得具有优良对映选择性 (96-97% ee)的手性产物2z-2ab。具有环外烯键的烯基底物1ac,能够获得90% ee的产物 2ac。接下来,作者进一步发现,具有薄荷醇(menthol)、冰片醇 (borneol)、金刚烷 (adamantane)、香叶醇 (geraniol)以及松油醇 (terpineol)结构单元的烯基底物,同样能够较好地参与上述的不对称氢化过程,并获得相应的手性产物2ad–2ah (95-97% ee)。进而表明这一全新的对映选择性氢化方案在具有天然产物以及药物结构单元的烯基底物的后期官能团化过程中具有潜在的应用价值。然而,将上述最佳的反应条件应用于非芳基1,1-二取代烯基底物的不对称氢化反应时,则无法获得优良的反应转化率与对映选择性 (42%转化率,54% ee)。

(图片来源:J. Am. Chem. Soc.)
接下来,该小组进一步考察上述不对称氢化策略的合成应用价值。首先,作者发现,在3 mol % 铁配合物存在下进行的克级规模实验中,能够获得98%收率以及98% ee的产物2a (Scheme 4a)。同时,作者观察到,在具有更高立体位阻的Lb·FeCl2催化剂存在的条件下,带有两种不同类型的碳-碳双键的底物1aj,同样能够获得95%反应收率与93% ee的产物2aj。并且,底物中的三取代烯键未参与相应的氢化过程 (Scheme 4b)。这一独特的底物识别性能在序列合成策略的设计中具有潜在的应用价值。

(图片来源:J. Am. Chem. Soc.)
为阐明上述不对称氢化过程的反应机理,作者进行一系列氘标记实验研究 (eq 1-4)。首先,作者发现,采用α-D-烯基化合物 d1–1a在上述标准反应条件下参与的不对称氢化过程,能够获得无氘原子置乱的产物d1–2a (eq 1)。这一事实表明,不对称氢化过程中,未涉及α-亚甲基的参与。而在d2–1a转化为d2–2a产物的过程中,能够观察到在烯键的末端位置,出现氘原子的少量丢失 (eq 2)。


接下来,作者观察到,采用D2参与的不对称转化过程中,产物d3–2a的苄基与甲基位置存在少量的氢原子 (eq 3)。而选择氘代氢硅烷参与相关反应时,产物d4–2a中的苄基与甲基位置能够观察到少量氘原子的存在 (eq 4)。上述结果表明,氢硅烷可能作为一种潜在的氢化物源,参与相应的催化循环过程。之后,该小组在D-氢硅烷与氢气之间的反应中,观察到H-D交换现象。进而表明,在催化过程中,活性铁中间体可能与氢硅烷以及H2之间经历氧化加成或σ-键复分解步骤,转化为另外一种具有催化活性的铁中间体 (eq 5)。


同时,在采用固体腈 (例如3-甲氧基苯腈)作为添加剂代替MeCN时,作者发现,反应同样能够顺利进行,在反应完成之后,能够回收相应的固体腈添加剂 (eq 6)。其它的机理实验,参阅Supporting Information。

根据氘标记实以及前期的文献研究[7],作者提出一种可能的反应机理(Scheme 5)。首先,在NaBHEt3活化剂存在下,通过手性铁配合物,形成铁氢化物中间体 A。之后,通过烯基化合物的配位与插入过程,形成1,2-插入的铁烷基物种 B。铁烷基物种 B与氢硅烷之间经历σ-键复分解过程,获得手性烷基产物与铁硅基物种 C,C与氢分子之间通过进一步的σ-键复分解过程,形成铁氢化物中间体 A。然而,目前仍无法排除铁烷基物种 B与氢分子之间的σ-键复分解机理路径。其中,乙腈添加剂的作用可能是抑制2, 1-插入过程。并且,乙腈添加剂的存在,同样可能抑制通过2, 1-插入形成的铁中间体通过β-氢消除,产生三取代烷基化合物的反应过程。

(图片来源:J. Am. Chem. Soc.)
总结
综上所述,浙江大学的陆展团队设计出一种全新的手性N,N,N-三齿配体及其铁配合物,进而首次实现铁催化剂促进的一系列烯基化合物的高度对映选择性氢化反应方法学。这一全新的不对称氢化策略中,采用氢硅烷与乙腈作为有效的添加剂能够提高反应活性与不对称氢化过程的选择性。同时,该小组通过氘标记实验,对相关的反应机理进行初步研究。
参考文献
[1] C. S. Shultz, S. W. Krska, Acc. Chem. Res. 2007, 40, 1320. doi: 10.1021/ar700141v. [2] S. Zhu, Q. Zhou, Acc. Chem. Res. 2017, 50, 988. doi: 10.1021/acs.accounts.7b00007. [3] H. Zhong, M. Shevlin, P. J. Chirik, J. Am. Chem. Soc. 2020, 142, 5272. doi: 10.1021/jacs.9b13876. [4] I. Bauer, H.-J. Knölker, Chem. Rev. 2015, 115, 3170. doi: 10.1021/cr500425u. [5] Y. Sunada, H. Ogushi, T. Yamamoto, S. Uto, M. Sawano, A. Tahara, H. Tanaka, Y. Shiota, K. Yoshizawa, H. Nagashima, J. Am. Chem. Soc. 2018, 140, 4119. doi: 10.1021/jacs.8b00812. [6] L. Zhang, Z. Zuo, X. Wan, Z. Huang, J. Am. Chem. Soc. 2014, 136, 15501. doi: 10.1021/ja5093908. [7] M. R. Friedfeld, M. Shevlin, G. W. Margulieux, L.-C. Campeau, P. J. Chirik, J. Am. Chem. Soc. 2016, 138, 3314. doi: 10.1021/jacs.5b10148.本文作者:杉杉
导读
烯烃的不对称氨基叠氮化(aminoazidation)和双叠氮化(diazidation)是一类构建手性含氮化合物的有效策略。近日,中科院福建物构所鲍红丽课题组在Angew. Chem. Int. Ed. 上发表论文,报道了铁催化苯乙烯分子间的不对称氨基叠氮化和双叠化反应,该反应仅使用较低催化量的催化剂和手性配体,通过自由基机理进行,涉及无环自由基的立体控制(可能通过基团转移过程)。
Iron-Catalyzed Radical Asymmetric Aminoazidation and Diazidation of Styrenes
Hongli Bao, Daqi Lv, Qiao Sun, Huan Zhou, Liang Ge, Yanjie Qu, Taian Li, Xiaoxu Ma, and Yajun Li
Angew. Chem. Int. Ed. ASAP DOI: 10.1002/anie.202017175

正文
对映体富集的邻二胺化合物广泛的存在于天然产物、药物和生物活性分子中,也可作为手性配体参与不对称催化。虽然可通过醛亚胺[1]和/或亚胺[2]还原偶联合成手性二胺化合物,但烯烃或二烯的催化不对称胺化反应作为最直接且便捷的方法(Scheme 1),更加受到科学家们的关注。如Shi[3]、Du[4]、Gong[5]、Muñiz[6]和Denmark[7]等分别报道了钯、碘、有机硒催化等催化烯烃或二烯的分子间二胺化反应。Michael[8]、Chemler[9]、Zeng[10]和Liu[11] 也报道了通过一种外部胺化试剂和烯烃衍生的底物,经钯或铜催化实现分子间/内的二胺化反应,从而合成手性吲哚和吡咯烷化合物。这其中烯烃的氨基叠氮化和双叠氮化可作为互补的方法来合成邻二胺化合物。此外,有机叠氮化物也作为一种通用反应底物,可进行多种反应,如Curtius重排、Regitz重氮转移、Aza-Wittig反应、Boyer重排等。目前文献中[12]仅有一例关于应用不对称氨基叠氮化反应来合成不同的取代哌啶的实例(Scheme 1c)。对于不对称氨基叠氮化/双叠氮化反应(基于无环体系的分子间反应),仍具有挑战。前期,有文献报道了金属催化自由基叠氮化反应的机理[13],通常涉及分子间基团转移机理。本课题组根据前期对铁催化烯烃的碳叠氮化反应的研究[14],也提出了叠氮基团转移机理。受Ready等报道的对映选择性Kharasch加成反应 [15a]、Zhang等报道的对映选择性分子间自由基C-H胺化反应[15d]以及本课题组[15c]和Liu[15e]报道的苯乙烯和丙烯酰胺的不对称碳叠氮化反应的启发,在此,本课题组将进一步报道一种铁催化苯乙烯的不对称氨基叠氮化和双叠氮化反应。


首先,作者以苯乙烯1a、NFSI(氮自由基前体)和TMSN3(叠氮源)为模型底物,进行了相关反应条件的筛选(Table 1)。反应的最佳条件为:以Fe(OTf)2 为催化剂,L5为配体,可在CHCl3溶剂中室温反应,获得96%收率和93:7 er的目标产物2。



在获得上述最佳反应条件后,作者开始对苯乙烯的底物范围进行了扩展(Scheme 2)。在对氨基叠氮化反应的范围扩展时发现,一系列不同取代的苯乙烯(如各种给电子基或吸电子基)均可顺利反应,并获得良好收率和er的相应产物3–32。同时,除5、16和32外,大多数反应使用L4或L5,L4可获得更好的对映选择性。值得注意的是,若底物中同时含有乙烯基和烯丙基/炔基时,反应优先发生在乙烯基上,从而获得产物25和26。2-乙烯基苯并[b]噻吩也为合适的底物,可获得90%收率和88:12 er的产物33。此外,通过增加叠氮化试剂的量和催化剂的量,可实现1,3-二乙烯基苯和1-溴-3,5-二乙烯基苯的双叠氮化反应,获得中等非对映选择性和较高er的产物34和35。
其次,作者通过再次优化条件后发现,手性铁催化剂适用于二或三取代的苯乙烯底物。对于三取代苯乙烯底物,可获得高达97:3 er的产物37–49。对于苯乙烯底物,可获得中等收率产物50,但er较低。对于不对称苯乙烯底物,可获得含有两个不对称中心的产物51和52,每个异构体的er均为中等水平。对于对称的苯乙烯底物,可获得双叠氮化合物53–56。



为了进一步证明反应的实用性,作者对产物8进行了后期衍生化(Scheme 3)。首先,8可与末端或内炔烃、苯炔前体或乙酰丙酮进行点击反应(Click reactions),从而获得高收率且对映选择性不损失的三唑产物57–62。其次,8可在铟粉的MeOH中回流,获得79%收率的手性伯胺63。若使用P(OMe)3还原化合物8,可获得81%收率的手性磷酰胺64。除叠氮化物以外,化合物8可经n-Bu4NF进行单脱磺酰化,以定量收率得到产物65,若经进一步还原即可获得单保护的1,2-二胺66,收率为62%。66若经一锅炔丙基化/分子内[3+2]环加成反应,可获得三唑并吡嗪衍生物67。


为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 4)。首先,在标准反应体系中加入TEMPO,仅获得24%收率的产物8,并分离出14%收率的氨基氧化产物68(Scheme 4a)。当加入BHT时,反应完全被抑制(Scheme 4b)。当加入1,1-二苯基乙烯时,反应也被抑制,仅获得40%收率的产物8(Scheme 4c)。在自由基钟实验中,反应以32%的收率形成开环产物70,且未获得氨基叠氮化产物71(Scheme 4d)。这些结果表明,该反应可能涉及苄基自由基的参与。其次,为了确定催化活性物质,作者合成了两种配合物L2-Fe(II)OTf2•THF和L2-Fe(II)OTf2•CH3CN,反应结果表明,L2-Fe(II)OTf2配合物可以用作催化活性物质(Scheme 4e)。


最后,作者提出了一种可能的反应机理(Scheme 5)。首先,铁催化剂与配体的配位形成铁(II)配合物(A),可与TMSN3进行配体交换,得到叠氮化铁(II)配合物(B)。紧接着,NFSI和B经单电子转移形成叠氮化铁(III)配合物(C)和双磺酰胺自由基(D)。同时,配合物A也可与NFSI经单电子转移形成配合物E和自由基(D),E与TMSN3反应生成叠氮化铁(III)配合物(C)。将双磺酰胺自由基基(D)与苯乙烯进行加成形成苄基自由基(F)。最后,苄基自由基可与配合物C经基团转移机理,从而获得目标产物并再生活性催化剂A。

总结
中科院福建物构所鲍红丽课题组报道了第一个铁催化苯乙烯的不对称氨基叠氮化和双叠氮化反应,从而合成了一系列手性胺化有机叠氮化物和二叠氮化物,并可进一步转化为各种具有价值的含氮化合物。同时,该反应具有温和的反应、广泛的底物范围、良好的官能团耐受性等特点。此外,该反应涉及苄基自由基参与。
参考文献
[1] X. Shao, K. Li, S. J. Malcolmson, J. Am. Chem. Soc. 2018, 140, 7083-7087. [2] M. Zhou, K. Li, D. Chen, R. Xu, G. Xu, W. Tang, J. Am. Chem. Soc. 2020, 142, 10337-10342. [3] a) H. Du, W. Yuan, B. Zhao, Y. Shi, J. Am. Chem. Soc. 2007, 129, 11688-11689; b) H. Du, B. Zhao, Y. Shi, J. Am. Chem. Soc. 2008, 130, 8590-8591; c) H. Du, B. Zhao, W. Yuan, Y. Shi, Org. Lett. 2008, 10, 4231-4234. [4] X. Hu, Z. Cao, Z. Liu, Y. Wang, H. Du, Adv. Synth. Catal. 2010, 352, 651-655. [5] M.-S. Wu, T. Fan, S.-S. Chen, Z.-Y. Han, L.-Z. Gong, Org. Lett. 2018, 20, 2485-2489. [6] K. Muniz, L. Barreiro, R. M. Romero, C. Martinez, J. Am. Chem. Soc. 2017, 139, 4354-4357. [7] Z. Tao, B. B. Gilbert, S. E. Denmark, J. Am. Chem. Soc. 2019, 141, 19161-19170. [8] E. L. Ingalls, P. A. Sibbald, W. Kaminsky, F. E. Michael, J. Am. Chem. Soc. 2013, 135, 8854-8856. [9] a) B. W. Turnpenny, S. R. Chemler, Chem. Sci. 2014, 5, 1786-1793; b) F. C. Sequeira, B. W. Turnpenny, S. R. Chemler, Angew. Chem. Int. Ed. 2010, 49, 6365-6368; Angew. Chem. Int. Ed.2010, 122, 6509 –6512. [10] S. Fu, H. Yang, G. Li, Y. Deng, H. Jiang, W. Zeng, Org. Lett. 2015, 17, 1018-1021. [11] G. Qiu, Q. Wang, J. Zhu, Org. Lett. 2017, 19, 270-273. [12] X. Li, X. Qi, C. Hou, P. Chen, G. Liu, Angew. Chem. Int. Ed. 2020, 59, 17239-17244; Angew. Chem. Int. Ed.2020, 132, 17392 –17397. [13] a) X. Huang, T. M. Bergsten, J. T. Groves, J. Am. Chem. Soc. 2015, 137, 5300-5303; b) L. Zhang, S. Liu, Z. Zhao, H. Su, J. Hao, Y. Wang, Chem. Sci. 2018, 9, 6085-6090. [14] M. F. Chiou, H. Xiong, Y. Li, H. Bao, X. Zhang, Molecules 2020, 25, 1224. [15] a) B. Chen, C. Fang, P. Liu, J. M. Ready, Angew. Chem. Int. Ed. 2017, 56, 8780-8784; Angew. Chem. Int. Ed.2017, 129, 8906 –8910; b) Y. Zeng, M. F. Chiou, X. Zhu, J. Cao, D. Lv, W. Jian, Y. Li, X. Zhang, H. Bao, J. Am. Chem. Soc. 2020, 142, 18014-18021; c) L. Ge, H. Zhou, M.-F. Chiou, H. Jiang, W. Jian, C. Ye, X. Li, X. Zhu, H. Xiong, Y. Li, L. Song, X. Zhang, H. Bao, Nat. Catal. 2021, 4, 28-35; d) L. M. Jin, P. Xu, J. Xie, X. P. Zhang, J. Am. Chem. Soc. 2020, 142, 20828-20836; e) L. Wu, Z. Zhang, D. Wu, F. Wang, P. Chen, Z. Lin, G. Liu, Angew. Chem. Int. Ed. 2021, 60, 6997-7001; Angew. Chem. Int. Ed.2021, 133, 7073-7077.概要
过渡金属催化的导向碳氢键官能化反应由于其步骤经济性以及碳氢键在有机分子中的普遍存在的特性,展示出了广阔的应用前景。导向的直接碳氢键官能化主要通过环金属化的金属有机中间体进行,虽然环金属化反应多报道于后过渡金属,但是含有Csp2-铁和Csp3-铁键的铁碳环金属有机化合物也有过报道。E. Nakamura等人首次发现通过生成铁碳环中间体可以实现杂原子导向的C-H活化C-C键生成反应[1, 2]。铁作为地壳中储量最大的过渡金属元素,由于其大储量以及廉价无毒的特性,铁催化作为一种环境友好且可持续的催化剂[3, 4],在碳氢键活化中的运用近年开始受到了广泛的关注。目前该反应已经拓展到Csp2-H以及Csp3-H与亲核试剂以及亲电试剂[6, 7]的反应,展示出了潜在的发展潜力和应用前景。

基本文献
- [1] Norinder, J.; Matsumoto, A.; Yoshikai, N.; Nakamura, E. J. Am. Chem. Soc. 2008, 130, 5858. DOI: 10.1021/ja800818b
- [2] Yoshikai, N.; Matsumoto, A.; Norinder, J.; Nakamura, E. Angew. Chem., Int. Ed. 2009, 48, 2925. DOI: 10.1002/anie.200900454
- [3] Nakamura, E.; Yoshikai, N. J. Org. Chem. 2010, 75, 6061. DOI: 10.1021/jo100693m
反应机理
- 该类型的铁催化导向碳氢官能化反应与以往的很多自由基类型的铁催化反应不同,该类铁催化的碳氢键活化是通过与贵金属Pd,Rh等类似的环金属化(cyclometallation)中间体进行。反应涉及到铁碳环的生成,以及铁碳环参与的还原消除或者氧化加成反应。由于铁金属有机中间体在分离上的困难,目前该反应的机理并不是十分明确。研究指出针对具体的情况“还原的低价铁”以及三价铁都可以作为活化碳氢键的活性物种。反应中活化碳氢键所使用的碱目前主要依赖于金属有机试剂这一类强碱。1,2-二氯代烷烃常作为该反应中氧化铁催化剂的氧化剂。

反应实例
- 铁催化的氮原子导向的直接碳氢键芳基化反应[1]

- 铁催化的非自由基C(sp3)-H直接芳基化[5]

- 铁催化的直接碳氢活化烯丙基化(与亲电试剂直接偶联)[7]

实验步骤
以反应实例1为例。在零度的冰水浴中,在惰气保护下,往0毫摩尔的反应物的无水无氧四氢呋喃溶液中加入乙酰丙酮铁以及配体,得到反应物和催化剂的混合物。在另一个Schlenk反应管中将格式试剂与锌盐混合并搅拌20分钟。将预先混合的催化剂和底物的混合溶液转移到制备的锌试剂溶液中。最后往该反应混合物中加入1,2-二氯异丁烷,然后在冰水浴中搅拌16小时。反应结束后,通过加入酒石酸钠钾的饱和水溶液淬灭反应。通过柱层析分离得到反应产物。
实验技巧
由于该类反应使用到金属有机试剂,如格氏试剂和锌试剂,反应对于溶剂的干燥程度要求比较高。使用新制的金属有机试剂是更佳的选择,这是由于久置的金属有机试剂往往由于保管不当而含有金属氢氧化物,这会抑制铁催化剂的活性。
参考文献
- [4] Ilies, L.; Nakamura, E. Iron-Catalyzed Cross-Coupling Reactions, in The Chemistry of Organoiron Compounds; Eds. Marek, I.; Rappoport, Z.; John Wiley & Sons, Ltd.: Chichester, UK, 2014.
- [5] Shang, R.; Ilies, L.; Matsumoto, A.; Nakamura, E. J. Am. Chem. Soc. 2013, 135, 6030. DOI: 10.1021/ja402806f
- [6] Matsubara, T.; Asako, S.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2014, 136, 646. DOI: 10.1021/ja412521k
- [7] Asako, S.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2013, 135, 17755. DOI: 10.1021/ja4106368
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
Alois Fürstner (1962年xx月xx日-) 是德国有机化学家、Max-Planc研究所(http://www.kofo.mpg.de/en)主任研究员(http://www.kofo.mpg.de/en/research/organometallic-chemistry)。
- 经历
1987 Technical University Graz 博士号取得 (H. Heidmann教授)
1990 Geneva大学 博士研究员 (W. Oppolzer教授)
1992 Technical University Graz
1993 多特蒙德大学 讲师
1993 Max・Planc 研究所 主任研究员
- 获奖经历
2000 Thieme-IUPAC prize
2002 阿瑟・C・科普学者奖
2005 Mukaiyama Award
2006 Otto Bayer Prize
- 研究
新型金属催化反应的开发及其在生理活性物质合成中的应用
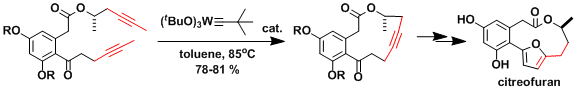
炔烃的复分解反应(Alkyne Metathesis)、研究通过铁催化的Kochi-Furstner偶联反应,并用其来开发独特的合成方法论。 (以下是citreofuran的合成实例例[1])。
- 评论&其他
- 相关文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!